

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Destacado (14)
Similar a Ictericia
Similar a Ictericia (20)
Más de junior alcalde
Más de junior alcalde (20)
Último
Último (20)
Ictericia
- 1. ICTERICIA
- 2. ANATOMIA HEPÁTICA Estómago Intestino delgado Intestino grueso Vesícula biliar HÍGADO Bazo
- 6. EP = Espacio porta; AH = Rama portal de la arteria hepática; VP = Rama de la vena porta; CB = Conductillo biliar; VC = Vena centrolobulillar; S = Sinusoide; T = Trabécula de células hepáticas LOBULILLO HEPÁTICO CB
- 7. Vena centrolobulillar Canal portal LOBULILLO HEPÁTICO
- 8. ACINO HEPÁTICO EP = Espacio porta; AH = Rama portal de la arteria hepática; VP = Rama de la vena porta; CB = Conductillo biliar; VC = Vena centrolobulillar; S = Sinusoide; T = Trabécula de células hepáticas
- 9. Diagrama que muestra la distribución de los lobulillos y acinos en el hígado Espacio porta Lobulillo Vena central Acino
- 14. METABOLISMO DE LA BILIRRUBINA
- 18. Bilirrubina conjugada Bilirrubina conjugada Bilirrubina libre Urobilinógenos Circulación enterohepática Vena porta Urobilinógeno fecal o Estercobilinógeno 40 - 280 mg/24h Acción bacteriana B-Glucuronidasa Mesobilifucsina COLOR MARRÓN DE HECES Captación – Transporte – Conjugación – Excreción Circulación Sistémica ( < 3mg/100mg) Urobilinógeno urinario 0 - 4 mg/24h Filtración y excreción Intestino Riñón Hígado
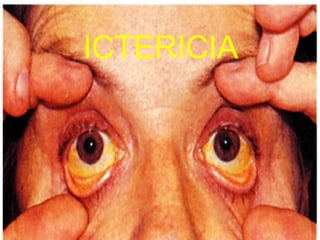
- 19. INTRODUCCIÓN A LA ICTERICIA Acumulación de bilirrubina en la sangre Pigmentación amarillenta de la piel y mucosas visibles, además del suero
- 20. Diagnóstico clínico diferencial Coloración amarillenta de mucosas y escleróticas ICTERICIA Piel toma coloración amarillenta PSEUDOICERICIA NUNCA ESCLERÓTICAS NI MUCOSAS * Drogas que producen impregnación cutánea: QUINACRINA * Uso excesivo de alimentos ricos en carotenos en individuos con deficiencia enzimática de carotenasa
- 22. ICTERICIA POR HIPERBILIRRUBINEMIA INDIRECTA NO CONJUGADA
- 23. ICTERICIA POR HIPERBILIRRUBINEMIA CONJUGADA
- 25. HEPATOCELULARES OBSTRUCTIVAS HEMOLÍTICAS CLASIFICACIÓN A CLASIFICACIÓN B ICTERICIA POR HIPERBILIRRUBINEMIA INDIRECTA NO CONJUGADA ICTERICIA POR HIPERBILIRRUBINEMIA CONJUGADA
- 36. ICTERICIAS HEMOLÍTICAS EXTRACORPUSCULARES INTRACORPUSCULARES ISOINMUNE NO ISOINMUNE INCOMPATIBILIDAD METERNO - FETAL INFECCIONES PARASITARIAS INCOMPATIBILIDAD GRUPOS SANGUINEOS POLICITEMIAS PYRCINASA Pyr DHG HEMOGLOBINOPATIAS DESTRUCCIÓN DE ERITROCITOS ANEMIA > ICTERICIA
- 39. ICTERICIAS PREHEPÁTICAS HEPÁTICAS POSHEPÁTICAS Hepatitis Cirrosis
- 42. * Se va a producir: -Infiltración de los espacios porta e interlobulillares por linfocitos y después por macrófagos. -Necrosis de hepatocitos. -Hiperplasia de las células de kupffer. -Grados variable de colestacia. -En la hepatitis leve, las zonas de necrosis se mantienen aisladas; en las formas que evolucionan a la cronicidad las necrosis confluyen y unen las venas centrolobulillares.
- 48. Infección aguda Enf. Subclínica Hepatitis Aguda Portador sano Infex persistente Hepatitis Crónica Recuperación Hepatitis fulminante Muerte Recuperación Cirrosis Carcinoma hepatocelular Muerte 60-65% 20-25% 5-10% 4% 10-33% 67-90% 99% 100% <1% 10% 20 - 50 %
- 51. Infección aguda Resolución Hepatitis crónica Hepatitis fulminante Enfermedad estable Cirrosis Carcinoma hepatocelular Cirrosis estable Muerte 15% 85% raro 80% 20% 50% 50%
- 59. Patogenia Proceso patológico central es la fibrosis progresiva y reorganización de la microarquitectura vascular del hígado Depósito de colágenos (tipos I y III) en el lobulillo Cirrosis Tractos fibrosos finos o gruesos. Canales vasculares nuevos en los tabiques que comunican las estructuras vasculares de la región portal con las venas hepaticas terminales Cortocircuitos por los que la sangre evita el parénquima Se forman estableciendo Creando
- 60. Depósito continuo de colágeno en el espacio de Disse del parénquima conservado Pérdida de las fenestraciones de las células endoteliales de los sinusoides. Cél. estrelladas perisinusoidales -Una potente actividad mitótica en áreas donde se desarrolla nueva fibrosis parenquimatosa. -Una variación transicional a miofibroblastos. -Aumento en la capacidad de síntesis y secreción de matriz extracelular. Aparición de las miofibrillas RV en el interior del parénquima hepático la luz de los canales vasculares sinusoidales. Se asocia a Sumado a Citocinas Estimulan Se activan
- 61. A través de éste proceso de lesión y fibrosis hepática Los hepatocitos restantes a regenerar y proliferar como nódulos dentro de los límites impuestos por los tabiques fibrosos. Hígado fibroso y nodular Alteración de la interfase entre el parénquima y los espacios porta Ictericia Obliteración de los conductillos biliares Son estimulados Resultado final y
- 64. Proceso existe impedimento en el normal flujo de bilis desde el polo canalicular del hepatocito hasta duodeno. COLESTASIS
- 74. Coloracion amarilla de piel mucosas y Esclerotica EXPRESIÓN CLÍNICA DE LA COLESTASIS ICTERICIA
- 75. Aparecer superficies extensión están en relación nivel de lípidos circulantes Dislipemia EXPRESIÓN CLÍNICA DE LA COLESTASIS XANTOMAS
- 78. CLASIFICACIÓN
- 81. COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS LITIASIS BILIAR
- 82. Colecistitis aguda Inflamación aguda de la vesícula biliar producida en la mayoría de los casos por obstrucción del cuello de la vesícula o del conducto cistico. COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 84. Inflamación mecánica Inflamación química Inflamación bacteriana presión intraluminal y distensión Fosfolipasa acción sobre la lecitina Lisolecitina Escherichia Coli Klebsiella Streptococcus Clostriduin Isquemia de la mucosa y pared vesicular
- 85. Edema inflamatorio Vías biliares y ganglios linfáticos Síndrome de Mirizzi Calculo impacta en el conducto cistico o en el cuello de la vesícula Compresión del conducto colédoco ICTERICIA COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 86. PANCREATITIS AGUDA Es un grupo de lesiones reversibles e irreversibles que se caracterizan por una inflamación del páncreas edema y necrosis necrosis del parénquima con hemorragia COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 87. EDEMA DE LA CABEZA DEL PANCREAS COMPRIME LA PORCION INTRAHEPATICA DEL CONDUCTO COLEDOCO ICTERICIA COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 88. ATRESIA BILIAR Es una obstrucción completa de la luz del árbol biliar extrahepatico dentro de los 3 meses de vida Forma fetal Desarrollo aberrante intrauterino del árbol biliar Forma perinatal Destrucción del árbol biliar después del nacimiento ICTERICIA COLESTASIS EXTRAHEPÁTICAS BENIGNAS
- 89. ATRESIA BILIAR
- 90. QUISTES DEL COLEDOCO Son dilataciones congénitas del conducto biliar común Porción libre Quiste Obstrucción del conducto Porción intraduodenal Divertículo Reflujo crónico del jugo pancreático hacia el árbol biliar Inflamación y estenosis de los conductos extrahepaticos ICTERICIA COLESTASIS EXTRAHEPÁTICAS NO NEOPLAICAS
- 91. Hospedador intermediario (Ingestión) Disgregación del embrióforo (Hexacanto) Penetración de vasos sanguíneos (Ganchos) Sistema porta Hidatidosis hepática COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 92. Hidatidosis hepática Quiste en el hígado Compresión del conducto biliar (Crecimiento de larva) Fuga de liquido del quiste hacia el árbol biliar Obstrucción biliar (total o parcial) ICTERICIA COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS Rotura Prolongada Angiocolitis, cirrosis
- 93. HEMOBILIA HIGADO Lesión traumática Lesión quirúrgica Absceso Aneurisma de la art. hepática Tumor benigno Hemorragia de la vías biliares. ICTERICIA COLESTASIS EXTRAHEPÁTICAS NO NEOPLASICAS
- 95. Carcinoma pancreático (60 – 80 a) Invasivo Reacción no neoplásica (Fibroblastos, linfocitos) Cabeza del páncreas Obstrucción del colédoco. Dilatación del árbol biliar ICTERICIA COLESTASIS EXTRAHEPÁTICAS NEOPLASICAS
- 97. Masas tumorales en espacios porta intrahepáticos Tumor nodular masivo Metástasis extrahepáticas Invasión linfática. Invasión vascular ICTERICIA Obstrucción del Conducto hepático común Colangiocarcinoma COLESTASIS EXTRAHEPÁTICAS NEOPLASICAS
- 100. Pancreatitis crónica ESTEATORREA Colestasis DOLOR DE INICIACIÓN PROGRESIVA Colangitis por obstrucción biliar complicada con infección TRIADA DE CHARCOT: Fiebre, dolor abdominal e ictericia Colestasis intra o extrahepatica COLURIA Colestasis biliar primaria PRURITO Obstrucción de la vía biliar por neoplasia ACOLIA PATOLOGIA ICTERICIA
- 101. ANAMNESIS
- 104. Tipo Cólico obstrucción por cálculos Dolor localizado en CSD hepatitis (vírica o tóxica) cirrosis y tumor Crónico e intenso carcinoma de páncreas Gástrico , irradiado a espalda enfermedad pancreática FIEBRE colangitis con colestasis extrahepática CON ESCALOFRIOS colangitis ,hepatitis vírica, origen farmacológico o etílico o leptospirosis DOLOR ABDOMINAL Dolor articular y cefaleas : hepatitis B (víricas) Diarrea : hepatitis vírica * acción laxante de los ácidos grasos no absorbidas Perdida de peso: carcinoma y tumores
- 105. SINTOMAS ACOMPAÑANTES color de la orina El mismo de siempre Hiperbilirrubinemia no conjugada “ coca cola” COLURIA BD ictericia hepatocelular u obstructiva sanguinolenta leptospirosis : icterohemorragica lesión hepática y renal * tóxicos color de las heces despigmentadas (ACOLIA) ictericia obstructiva En la coledocolitiasis la acolia suele ser intermitente, mientras que en las neoplasias habitualmente es continua. hiperpigamentadas ictericia hemolítica
- 106. Enfermedad hepatocelular . Anorexia, malestar general, mialgias, pródromos de tipo viral, exposición infecciosa conocida, Antecedentes de transfusiones, uso de drogas IV, alcohol, fármacos hepatotóxicos o antecedentes familiares de ictericia. Obstructiva . Dolor abdominal, fiebre, baja de peso, escalofríos,cirugía biliar previa
- 107. HEPATITIS HEPATITIS A HEPATITIS B HEPATITIS C Modalidad oral-fecal; transmitida por personas que preparan alimentos Mediante aguas negras; beber agua o comer alimentos contaminados con un comienzo insidioso, con anorexia, molestias abdominales vagas, náusea y vómito, a veces artralgia y erupciones, que puede culminar en ictericia. La fiebre puede ser leve o no presentarse. Hay casos fulminantes y mortales de necrosis hepática aguda. Contacto sexual . Vía perinatal (vertical) Exposición percutánea (IV,SC,ID) ,a través de las mucosas a los líquidos corporales infectantes: transfusión de sangre, uso compartido de agujas durante la inyección de drogas IV, hemodiálisis, acupuntura, tatuajes y pinchazos de aguja . fiebre,malestar general, anorexia, náusea y molestias abdominales, seguidas en pocos días de coloración amarillenta de la piel y mucosas (ictericia).(puede no haber) En la mayoría de los niños, la infección pasa inadvertida con pocos o ningún síntoma . No se conoce forma crónica de la enfermedad.
- 109. EXPLORACION FISICA
- 111. litiasis coledociana neoplasia pancreática Dolor agudo en hipocondrio derecho o epigastrio Dolor epigástrico de aparición tardía, constante e intenso que irradia a dorso Dolor abdominal Hepatomegalia dolorosa a palpación : hepatitis aguda o tumor de rápido crecimiento Vesícula palpable dolorosa : colesistitis aguda
- 112. Infiltración neoplásica de la cápsula hepatica y El peritoneo Roce en el área hepática Hepatitis .ICC con hepatomegalia aguda Hígado sensible a la palpación Necrosis hepatica aguda ,cirrosis postnecrotica Hígado pequeño Neoplasias , quistes,abcesos Nodulos hepaticos palpables Hepatoma , metastasis hepatica Higado de consistencia pétrea Trastorno intrahepatico , ictericia hemolitica Higado : tamano normal Anemia hemolitica, proceso maligno y Cirrosis Palidez SIGNIFICADO POSIBLE HALLAZGO Colangitis supurada Fiebre , escalofrios Cirrosis con trombocitopenia Púrpura
- 113. Excoriaciones COLESTASIS PROLONGADAS Y CIRROSIS BILIAR xantomas Acropaquia
- 114. Ascitis y venas periumbilicales dilatadas Cirrosis , hepatitis , hipertensión portal Cirrosis con circulación colateral Esplenomegalia Posible soplo venoso umbilical
- 115. HEPATOMEGALIA Cirrosis alcoholica , higado graso , colestasis , Hepatitis , congestión venosa ,proceso maligno, ICC , en sepsis (tifoide y brucella) .
- 116. Contractura de Dupuytren. La retracción de los tendones flexores palmares impide la extensión completa de los dedos. También :ginecomastia , Vello axilar y pubiano disminuida CIRROSIS ALCOHOLICA Parotidomegalia izquierda
- 117. ARAÑAS VASCULARES ERITEMA PALMAR HEPATOPATIA AGUDA O CRONICA
- 119. Signo de Murphy Colelitiasis coledocolitiasis Figura 3. Signo Murphy
- 121. LABORATORIO Y DIAGNÓSTICO TRASTORNOS DEL METABOLISMO DE LA BILIRRUBINA QUE PROVOCAN HIPERBILIRRUBINEMIA MIXTA O PREDOMINANTEMENTE CONJUGADA En la hiperbilirrubinemia producida por enfermedad hepática adquirida (p. ej. hepatitis aguda, cálculos en el colédoco) suele haber elevaciones de las concentraciones séricas tanto de bilirrubina conjugada como no conjugada. Aunque la obstrucción del árbol biliar o la lesión colestásica hepatocelular en ocasiones pueden presentarse con una hiperbilirrubinemia predominantemente conjugada, en general no es posible distinguir las causas de ictericia intrahepáticas de las extrahepáticas basándose en los valores séricos o las proporciones relativas de bilirrubina no conjugada y conjugada. La principal razón para determinar las cantidades de ambos tipos de bilirrubina en suero es hacer una diferenciación inicial de los trastornos parenquimatosos hepáticos y obstructivos (que cursan con hiperbilirrubinemia mixta conjugada y no conjugada) de los trastornos hereditarios y hemolíticos, antes expuestos, que se asocian con hiperbilirrubinemia no conjugada. DEFECTOS FAMILIARES EN LA FUNCIÓN EXCRETORA HEPÁTICA Síndrome de Dubin-Johnson Este trastorno benigno, más o menos raro, se caracteriza por una hiperbilirrubinemia predominantemente conjugada de baja intensidad (cuadro 284-2). Las concentraciones totales de bilirrubina generalmente se sitúan entre 34 y 85 mol/L (2 y 5 mg/100 ml), aunque en algunas ocasiones son normales o alcanzan valores de hasta 340 a 430 mol/L (20 a 25 mg/100 ml), y pueden fluctuar mucho en cada paciente. El grado de hiperbilirrubinemia puede aumentar por la presencia de enfermedades intercurrentes, anticoncepción oral y embarazo. Dado que la hiperbilirrubinemia se debe predominantemente a un incremento de la bilirrubina conjugada, por lo general hay bilirrubinuria. Aparte del incremento de los valores séricos de bilirrubina, el resto de los parámetros bioquímicos hepáticos son normales. La exploración física suele ser normal, excepto por la presencia de ictericia, aunque ocasionalmente hay hepatoesplenomegalia. Los pacientes con síndrome de Dubin-Johnson ( Dubin-Johnson syndrome , DJS) suelen encontrarse asintomáticos, aunque algunos refieren síntomas generales vagos. Es común que estos últimos pacientes ya hayan sido sometidos a extensos (y con frecuencia innecesarios) estudios diagnósticos por una ictericia no explicada, y presentan gran ansiedad. En las mujeres el trastorno puede ser subclínico hasta que se embarazan o empiezan a tomar anticonceptivos orales, momento en que la hiperbilirrubinemia bioquímica se convierte en ictericia manifiesta. Incluso en estas situaciones son normales el resto de las pruebas de función hepática, incluidas las de fosfatasa alcalina y actividades de transaminasas. Una característica esencial del síndrome de Dubin-Johnson es que en los lisosomas de los hepatocitos centrolobulillares se acumula un pigmento granular intensamente negro. Como consecuencia el hígado también puede verse negro a simple vista. Se cree que este pigmento procede de los metabolitos de la adrenalina que no se excretan normalmente. Puede desaparecer durante los brotes de hepatitis vírica, para volver a acumularse poco a poco después de la recuperación. En el síndrome de Dubin-Johnson está afectada la excreción biliar de una serie de compuestos aniónicos, como varios agentes colecistográficos, así como de la sulfobromoftaleína (Bromsulphalein, BSP), un colorante sintético utilizado antiguamente en una prueba de función hepática. En esta prueba se determinaba la tasa de desaparición de la BSP después de administrar un bolo intravenoso. La BSP se conjuga con glutatión en el hepatocito; en condiciones normales el conjugado resultante se excreta con rapidez en el canalículo. Los pacientes con síndrome de Dubin-Johnson muestran un incremento característico de su concentración plasmática 90 min después de la inyección, a causa del reflujo de la BSP conjugada hacia la circulación desde los hepatocitos. Colorantes como el verde de indocianina (ICG), que son captados por los hepatocitos pero que no sufren ninguna metabolización antes de su excreción biliar, no muestran este fenómeno de reflujo. Los estudios con infusión continua de BSP sugieren que existe una reducción del t máx de excreción biliar. En el síndrome de Dubin-Johnson es normal la eliminación de ácidos biliares, incluida la captación hepatocelular y la excreción biliar. Estos pacientes tienen concentraciones séricas y biliares normales de ácidos biliares, y no presentan prurito. Por analogía con lo observado en diversas cepas de ratas mutantes, se ha descubierto que el defecto selectivo en la excreción biliar de los conjugados de bilirrubina y de algunos otros compuestos orgánicos (pero no de los ácidos biliares) que caracteriza al síndrome de Dubin-Johnson refleja expresiones defectuosas de MRP2, un transportador de membrana canalicular dependiente de ATP. Varias mutaciones diferentes del gen MRP2 producen el fenotipo de Dubin-Johnson, que tiene un patrón de herencia autosómico recesivo. Aunque sin duda el MRP2 es importante en la excreción biliar de bilirrubina conjugada, el hecho de que el pigmento siga excretándose en ausencia del transportador sugiere que otras proteínas transportadoras, todavía desconocidas, intervienen de manera secundaria en este proceso. Los pacientes con síndrome de Dubin-Johnson también tienen una anomalía diagnóstica en la eliminación urinaria de coproporfirina. Existen dos isómeros naturales de coproporfirina, I y III. En condiciones normales, alrededor de 75% de la coproporfirina en orina es el isómero III. Sin embargo, en la orina de los pacientes con síndrome de Dubin-Johnson, aunque el contenido total de coproporfirina es normal, más de 80% es isómero I. Los heterocigotos para el síndrome muestran un patrón intermedio. La base molecular de este fenómeno sigue sin dilucidarse. Síndrome de Rotor Este trastorno autosómico recesivo benigno es clínicamente similar al síndrome de Dubin-Johnson, aunque se observa todavía con menos frecuencia (cuadro 284-2). Una de las principales diferencias fenotípicas es que el hígado de los pacientes con síndrome de Rotor no presenta aumento de la pigmentación y su aspecto es totalmente normal. La única anomalía en las pruebas habituales de laboratorio es un incremento de la bilirrubina sérica total debido a un aumento predominante de la bilirrubina conjugada. Esto se acompaña de bilirrubinuria. Existen otras características que diferencian los síndromes de Rotor y Dubin-Johnson. En el de Rotor, la vesícula biliar suele visualizarse con la colecistografía oral, al contrario de lo que sucede en el síndrome de Dubin-Johnson. El patrón de eliminación urinaria de coproporfirina también es diferente. El del síndrome de Rotor es similar al de muchos trastornos adquiridos de la función hepatobiliar, en los que la coproporfirina I, el principal isómero en la bilis, refluye del hepatocito hacia la circulación y se elimina por la orina. Así, en el síndrome de Rotor, la eliminación urinaria total de coproporfirina está muy incrementada, en contraste con los valores normales que se observan en el síndrome de Dubin-Johnson. Aunque la fracción de coproporfirina I está elevada en la orina, suele ser menor de 70% del total, en comparación con el 80% o más del síndrome de Dubin-Johnson. Ambas enfermedades pueden distinguirse también por sus patrones de excreción de BSP. Aunque el aclaramiento de la BSP del plasma en el síndrome de Rotor está retrasado, no hay reflujo de BSP conjugada hacia la circulación como se observa en el síndrome de Dubin-Johnson. Los análisis cinéticos de los estudios de infusión plasmática de BSP sugieren la presencia de un defecto en el almacenamiento intrahepatocelular de este compuesto. Esto nunca se ha demostrado directamente, y la base molecular del síndrome de Rotor sigue siendo desconocida
- 122. TRASTORNOS DEL METABOLISMO DE LA BILIRRUBINA QUE PROVOCAN HIPERBILIRRUBINEMIA MIXTA O PREDOMINANTEMENTE CONJUGADA PERFIL HEPATICO VALORES DE LABORATORIO FOSFATASA ALCALINA 60 – 240 UI/L. BILIRRUBINAS : Transaminasas GOT - ALT : Valor normal entre 0 y 37 U/L GPT - AST : Valor normal entre 0 y 41 U/L Bilirrubina directa: hasta 0,2 mg/dl Bilirrubina indirecta : hasta 0,8 mg/dl BILIRRUBINA TOTAL: hasta 1 mg/dl Albúmina Globulinas 3,5 a 4,8 g/dl 2,6 a 3,1 g/dl PROTEÍNAS TOTALES 6,1 a 7,9 g/dl En la hiperbilirrubinemia producida por enfermedad hepática adquirida (p. ej. hepatitis aguda, cálculos en el colédoco) suele haber elevaciones de las concentraciones séricas tanto de bilirrubina conjugada como no conjugada. Aunque la obstrucción del árbol biliar o la lesión colestásica hepatocelular en ocasiones pueden presentarse con una hiperbilirrubinemia predominantemente conjugada, en general no es posible distinguir las causas de ictericia intrahepáticas de las extrahepáticas basándose en los valores séricos o las proporciones relativas de bilirrubina no conjugada y conjugada. La principal razón para determinar las cantidades de ambos tipos de bilirrubina en suero es hacer una diferenciación inicial de los trastornos parenquimatosos hepáticos y obstructivos (que cursan con hiperbilirrubinemia mixta conjugada y no conjugada) de los trastornos hereditarios y hemolíticos, antes expuestos, que se asocian con hiperbilirrubinemia no conjugada. DEFECTOS FAMILIARES EN LA FUNCIÓN EXCRETORA HEPÁTICA Síndrome de Dubin-Johnson Este trastorno benigno, más o menos raro, se caracteriza por una hiperbilirrubinemia predominantemente conjugada de baja intensidad (cuadro 284-2). Las concentraciones totales de bilirrubina generalmente se sitúan entre 34 y 85 mol/L (2 y 5 mg/100 ml), aunque en algunas ocasiones son normales o alcanzan valores de hasta 340 a 430 mol/L (20 a 25 mg/100 ml), y pueden fluctuar mucho en cada paciente. El grado de hiperbilirrubinemia puede aumentar por la presencia de enfermedades intercurrentes, anticoncepción oral y embarazo. Dado que la hiperbilirrubinemia se debe predominantemente a un incremento de la bilirrubina conjugada, por lo general hay bilirrubinuria. Aparte del incremento de los valores séricos de bilirrubina, el resto de los parámetros bioquímicos hepáticos son normales. La exploración física suele ser normal, excepto por la presencia de ictericia, aunque ocasionalmente hay hepatoesplenomegalia. Los pacientes con síndrome de Dubin-Johnson ( Dubin-Johnson syndrome , DJS) suelen encontrarse asintomáticos, aunque algunos refieren síntomas generales vagos. Es común que estos últimos pacientes ya hayan sido sometidos a extensos (y con frecuencia innecesarios) estudios diagnósticos por una ictericia no explicada, y presentan gran ansiedad. En las mujeres el trastorno puede ser subclínico hasta que se embarazan o empiezan a tomar anticonceptivos orales, momento en que la hiperbilirrubinemia bioquímica se convierte en ictericia manifiesta. Incluso en estas situaciones son normales el resto de las pruebas de función hepática, incluidas las de fosfatasa alcalina y actividades de transaminasas. Una característica esencial del síndrome de Dubin-Johnson es que en los lisosomas de los hepatocitos centrolobulillares se acumula un pigmento granular intensamente negro. Como consecuencia el hígado también puede verse negro a simple vista. Se cree que este pigmento procede de los metabolitos de la adrenalina que no se excretan normalmente. Puede desaparecer durante los brotes de hepatitis vírica, para volver a acumularse poco a poco después de la recuperación. En el síndrome de Dubin-Johnson está afectada la excreción biliar de una serie de compuestos aniónicos, como varios agentes colecistográficos, así como de la sulfobromoftaleína (Bromsulphalein, BSP), un colorante sintético utilizado antiguamente en una prueba de función hepática. En esta prueba se determinaba la tasa de desaparición de la BSP después de administrar un bolo intravenoso. La BSP se conjuga con glutatión en el hepatocito; en condiciones normales el conjugado resultante se excreta con rapidez en el canalículo. Los pacientes con síndrome de Dubin-Johnson muestran un incremento característico de su concentración plasmática 90 min después de la inyección, a causa del reflujo de la BSP conjugada hacia la circulación desde los hepatocitos. Colorantes como el verde de indocianina (ICG), que son captados por los hepatocitos pero que no sufren ninguna metabolización antes de su excreción biliar, no muestran este fenómeno de reflujo. Los estudios con infusión continua de BSP sugieren que existe una reducción del t máx de excreción biliar. En el síndrome de Dubin-Johnson es normal la eliminación de ácidos biliares, incluida la captación hepatocelular y la excreción biliar. Estos pacientes tienen concentraciones séricas y biliares normales de ácidos biliares, y no presentan prurito. Por analogía con lo observado en diversas cepas de ratas mutantes, se ha descubierto que el defecto selectivo en la excreción biliar de los conjugados de bilirrubina y de algunos otros compuestos orgánicos (pero no de los ácidos biliares) que caracteriza al síndrome de Dubin-Johnson refleja expresiones defectuosas de MRP2, un transportador de membrana canalicular dependiente de ATP. Varias mutaciones diferentes del gen MRP2 producen el fenotipo de Dubin-Johnson, que tiene un patrón de herencia autosómico recesivo. Aunque sin duda el MRP2 es importante en la excreción biliar de bilirrubina conjugada, el hecho de que el pigmento siga excretándose en ausencia del transportador sugiere que otras proteínas transportadoras, todavía desconocidas, intervienen de manera secundaria en este proceso. Los pacientes con síndrome de Dubin-Johnson también tienen una anomalía diagnóstica en la eliminación urinaria de coproporfirina. Existen dos isómeros naturales de coproporfirina, I y III. En condiciones normales, alrededor de 75% de la coproporfirina en orina es el isómero III. Sin embargo, en la orina de los pacientes con síndrome de Dubin-Johnson, aunque el contenido total de coproporfirina es normal, más de 80% es isómero I. Los heterocigotos para el síndrome muestran un patrón intermedio. La base molecular de este fenómeno sigue sin dilucidarse. Síndrome de Rotor Este trastorno autosómico recesivo benigno es clínicamente similar al síndrome de Dubin-Johnson, aunque se observa todavía con menos frecuencia (cuadro 284-2). Una de las principales diferencias fenotípicas es que el hígado de los pacientes con síndrome de Rotor no presenta aumento de la pigmentación y su aspecto es totalmente normal. La única anomalía en las pruebas habituales de laboratorio es un incremento de la bilirrubina sérica total debido a un aumento predominante de la bilirrubina conjugada. Esto se acompaña de bilirrubinuria. Existen otras características que diferencian los síndromes de Rotor y Dubin-Johnson. En el de Rotor, la vesícula biliar suele visualizarse con la colecistografía oral, al contrario de lo que sucede en el síndrome de Dubin-Johnson. El patrón de eliminación urinaria de coproporfirina también es diferente. El del síndrome de Rotor es similar al de muchos trastornos adquiridos de la función hepatobiliar, en los que la coproporfirina I, el principal isómero en la bilis, refluye del hepatocito hacia la circulación y se elimina por la orina. Así, en el síndrome de Rotor, la eliminación urinaria total de coproporfirina está muy incrementada, en contraste con los valores normales que se observan en el síndrome de Dubin-Johnson. Aunque la fracción de coproporfirina I está elevada en la orina, suele ser menor de 70% del total, en comparación con el 80% o más del síndrome de Dubin-Johnson. Ambas enfermedades pueden distinguirse también por sus patrones de excreción de BSP. Aunque el aclaramiento de la BSP del plasma en el síndrome de Rotor está retrasado, no hay reflujo de BSP conjugada hacia la circulación como se observa en el síndrome de Dubin-Johnson. Los análisis cinéticos de los estudios de infusión plasmática de BSP sugieren la presencia de un defecto en el almacenamiento intrahepatocelular de este compuesto. Esto nunca se ha demostrado directamente, y la base molecular del síndrome de Rotor sigue siendo desconocida
- 123. No Hemólisis Hct >38 no evidencia de Hemólisis * Síndrome de Gilbert * Farmacos, drogas * Ingesta pobre de lipidos FOSFATASA ALCALINA FOSFATASA ALCALINA Ictérica por colestasis Medidas de AST/ALT ( A ) ( B ) Medida de Fosfatasa Alcalina Bilirrubina conjugada Bilirrubina no conjugada Medidas de bilirrubina ICTERICIA Frotis de sangre (Hct)
- 125. A) Ictericia por colestasis Determinar Extrahepático Intrahepatico ECOGRAFIA dilatación de los conductos SI NO INTRAHEPATICO EXTRAHEPATICO CONFUSIONES Cirrosis Colangitis esclerosante