Immunohistochemistry
•Télécharger en tant que PPT, PDF•
39 j'aime•16,963 vues

Follow Atifa Ambreen: Youtube: https://www.youtube.com/channel/UCVTn-4KeMtXKy1h3_9JbVmw Facebook: https://www.facebook.com/atifaambreen786/ Instagram: https://www.instagram.com/atifa.ambreen/ https://www.instagram.com/invites/contact/?i=1qo1my1nb2qfx&utm_content=kbo8bi7 Snapchat: https://www.snapchat.com/add/atifa.ambreen Twitter: https://twitter.com/atifa_ambreen SUBSCRIBE ... . Email for inquiries: atifa.ambreen@gmail.com

Recommandé
Contenu connexe
Tendances
Tendances (20)
Similaire à Immunohistochemistry
Similaire à Immunohistochemistry (20)
Plus de Atifa Ambreen
Plus de Atifa Ambreen (20)
Dernier
Dernier (20)
Immunohistochemistry
- 2. Immunohistochemistry • What is it? • What can it tell you? • What are its limitations? • How is it done? • What are the controls? • Quantification • Single vs double or triple labelling
- 3. Antibodies and Antigens www.IHCworld.com Fab region Paratope + Epitope Ab-Ag complex
- 4. What is immunohistochemistry? – Immunohistochemistry is the localization in tissues of an (known) antigen by means of antibodies directed towards that (specific) antigen. – Ab-Ag complex is visualised using a marker. – Immunocytochemistry refers to localization in isolated cells or labelling directed to cell specific compartment (e.g. the cell membrane, Golgi or lysosomes, etc.).
- 5. Immunohistochemistry • What can it tell you? – Tissue or cellular localization of an antigen with cellular or subcellular resolution. – Specific cell or tissue expression of an antigen. – Major (but probably not minor) changes in the expression pattern of an antigen. – Dynamic changes in the expression of an antigen; particularly useful in animal models of disease
- 6. Immunohistochemistry • What are its inherent limitations? – Virtually nothing about quantity of antigen. – Despite doing rigorous controls, it cannot really tell you that an antigen is in the tissue. It can only show “immunoreactivity” or “like- immunoreactivity”. This distinction is often overlooked.
- 8. How is IHC done? • Preparation of Ab’s • Preparation of tissue – Fixation – Presentation – Chemical preparation • 1° Ab application • Visualisation of 1° Ab
- 9. Preparation of antibodies • Largely commercial • Often of spurious quality and variable titres • Rarely well-characterized – Let the literature be a guide, but do not hesitate to telephone or email potential sources or people in the field to get advice. • Polyclonal – Ag injected into host animal. Serum collected and purified. – Multiple antibodies produced by different cell types bind multiple epitopes on Ag • Monoclonal – Ag injected into mouse. Lymphocytes isolated, hybridized. – One antibody produced by one cell type binds one epitope on Ag.
- 10. Preparation of tissue - fixation Prevention of Ag translocation and/or degradation • Lots fixatives and they ALL potentially alter the presentation of antigen, because they crosslink protein and/or carbohydrates in the cell. • Most common fixatives are aldehydes (formalin, paraformaldehyde, glutaraldheyde (EM), Zamboni’s fixative). • Alcohols and acetone can be used, but they precipitate proteins and so tend to be good for cell-surface antigens. • Fixation can be performed at the level of the animal, tissue samples, e.g. biopsies, or after sectioning or mounting of fresh tissues or cells on a slide or in a dish.
- 11. Preparation of tissue - fixation • Whole animal – Perfuse via vasculature – typically intracardiac. – ADVANTAGES: – Removes blood – Ag degradation prevented before dissection – improves specimen quality – Effective fixation – All tissues fixed – DISADVANTAGES: – Time consuming – Messy – Safety concerns – use fume hood – Uses a lot of fixative
- 12. Preparation of tissue - fixation • Tissue specimens – Immersion fixation. – ADVANTAGES: – Quick, easy and safe – Excellent for small samples – Can prepare and manipulate tissue prior to fixation eg flat mounts of gut tissue – DISADVANTAGES: – Blood present – Large samples may take long time – not effectively fixed
- 13. Preparation of tissue - fixation • Cell preparations, fresh-frozen tissues mounted onto slides – Immersion fixation. – ADVANTAGES: – Quick, easy and effective – Fast for diagnostics – Good for cell surface markers – Rapid fixation – DISADVANTAGES: – Fresh frozen tissues need to be handled by skilled technician as morphology can suffer and degradation can occur
- 14. Preparation of tissue - embedding • Embedding - Paraffin or Cryostat – Preserve tissue integrity when sectioning – Paraffin infiltrates tissues – Cryo-embedding requires cryoprotection – Sucrose – Prevents ice crystals forming before embedding and freezing
- 15. Preparation of tissue - sectioning • Thickness main consideration – confocal microscopy revolutionised visualisation in thicker tissues • Sectioning: • Sledge microtome – paraffin • Cryostat – frozen • Vibratome – thicker tissues don’t require embedding • Whole mount: • Flat mount – no sectioning • Useful for visulaisation laminar structures eg enteric nervous system
- 16. Preparation of tissue • Improving antibody penetration – Reduce crosslinking – Reduce membrane barriers – Improve the presentation of the antigen • Antigen retrieval for paraffin embedding – Heat and/or proteolysis • DMSO/Ethanol washes • Triton X or other detergents to reduce surface tension therefore use less reagent
- 17. Blocking steps • Blocking unwanted tissue antigens – Improves signal to noise by preventing Fc receptor binding – Use normal serum of host species of the secondary antibody prior to 1° Ab incubation to reduce hydrophobic interaction of cross-linked proteins and Ab • Blocking unwanted endogenous peroxidase – Visualise with DAB substrate – Eliminate with hydrogen peroxide
- 18. Antibody application • Incubation with primary antibody – Floating immersion – On slides on a stage in humid chamber • Conditions should include consideration that they degrade • Use the lowest effective dilution, which needs to be determined empirically
- 19. Direct immunofluorescence • One step • Ab is labeled with fluorescent tag • Technique is fast • Low sensitivity due to lack of signal amplification
- 20. Indirect immunofluorescence • Two steps • Step 1 – bind 1° Ab to Ag • Step 2 – bind fluorescent labeled 2° Ab to 1° Ab • Sensitive – signal amplification • Use one 2° Ab for many unlabeled 1° Ab
- 21. Indirect immunoenzyme • Peroxidase anti-peroxidase or PAP method • Three steps • Step 1 – bind 1° Ab to Ag • Step 2 – bind unconjugated 2° Ab to 1° Ab • Step 3 – bind PAP complex to 2° Ab • Sensitive – signal amplification • Use DAB as a substrate for peroxidase colourimetric end product http://www.biologie.uni-regensburg.de/Zoologie/Schneuwly/Hofbauer/DROSI/strentw42.htm
- 22. Indirect immunoenzyme • Avidin-Biotin-Complex or ABC method • Three steps • Step 1 – bind 1° Ab to Ag • Step 2 – bind biotinylated 2° Ab to 1° Ab • Step 3 – bind avidin-biotin- peroxidase complex to 2° Ab • Very sensitive – signal amplification • Use DAB as a substrate for peroxidase colourimetric end product http://www.biologie.uni-regensburg.de/Zoologie/Schneuwly/Hofbauer/DROSI/strentw42.htm
- 23. Capturing images • Take convincing images!! • If the picture isn’t good you may as well have not done the experiment. • Consider composition and make sure you take representative images. • DO NOT MANIPULATE IMAGES
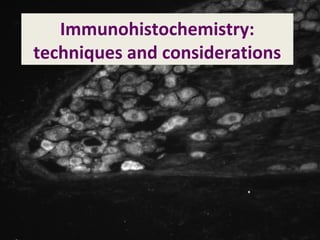
- 24. PGP9.5 immunoreactivity in rat nodose ganglia
- 25. Controls • Method specificity – Controls for tissue, reagents, secondary antibodies, autofluorescence, non-specific fluorescence, endogenous enzymes, etc., etc. • APPROACH - Leave out the primary antibody, do everything else the same. • Negative control – no staining – if you see something there is a problem. • IT IS NOT A CONTROL FOR THE SPECIFICITY OF THE LABELLING THAT YOU DO SEE!!
- 26. Controls • Antibody specificity – Controls for the specificity of the reaction • APPROACHES 1. Preabsorb the primary antibody with the immunizing antigen (question a company which does not supply this) 2. Replace the primary with preimmune serum from the same animal that the primary was raised in – or control ascites fluid for monoclonals • Negative control – no staining – if you see something there is a problem. • IT IS A CONTROL FOR THE SPECIFICITY OF THE LABELLING THAT YOU SEE – BUT – IT DOES NOT SAY THIS IS THE REAL ANTIGEN.
- 27. PPARa immunoreactivity in rat ileum PPARa antibody preabsorbed in blocking peptide in rat ileum
- 28. Controls • Antibody specificity – Positive controls • APPROACHES 1. Label tissues and/or transfected cells that are known to express the antigen of interest • Can be very good for well-established antigens, but is limited for novel antigens • Strongly encouraged as it is “positive”
- 29. Controls • Antibody specificity – KO MICE – a new approach that is gaining popularity • APPROACHES 1. Label tissues in mice with and without the gene product of interest 2. No labelling in the KO mouse is very powerful and speaks highly to the specificity of the reagents • Great if you can do it. • Note that not all gene products are actually knocked out fully.
- 30. Quantification in immunohistochemistry • Know the limits of the technique! • Can readily define labelled area • Can readily define the number of labelled cells if you can count total cells or some subset of cells • Much harder to define amount of labelling and this should be avoided unless strictly necessary and then only when carefully performed. Note that linearity of a signal is hard to prove.
- 31. GFAP expression is enhanced in colitis in GFP-GFAP transgenic mice
- 32. Single vs Double or Triple labelling • Sophisticated and fraught with difficulties that are not always appreciated. – How to incubate primary antibodies • Sequential vs combined – Interactions with secondary antibodies • Additional controls are required – “Bleed through” of fluorochromes • Need to assess filters • Consider sequential vs concurrent scanning when using a confocal microscope • If one reaction is very intense it can “bleach” a much weaker one in an overlay
- 33. Single vs Double or Triple labelling – Consider the species and choice of reagents carefully. • Must test specificity of secondary antibodies – cannot assume they not cross-react. Rigorous testing is recommended. – Primary A vs Secondary A – Primary B vs Secondary A – Primary B vs Secondary B – Primary A vs Secondary B – Primary A + Primary B vs Secondary A – Primary A + Primary B vs Secondary B – Primary A + Primary B vs Secondary A + Secondary B – Primary A + Secondary A + Secondary B – Primary B + Secondary A + Secondary B
- 34. PPARa immunoreactivity is localised to the nuclei of neurons in rat ileum
- 35. Neurobiotin Calbindin Merged image 50µm Intrinsic primary afferent neurons of the myenteric plexus are Dogiel Type II neurons that express calbindin
- 36. IL-1β activates neurons and glia in the submucosal plexus of the ileum.
- 37. Red (CY3): rat anti-substance P Green (FITC): mouse anti-VIP Blue (AMCA): rabbit anti-CGRP Distribution of nerves in the guinea pig ileum submucosal plexus TRIPLE LABELLING
- 38. Blue - DAPI; Green (FITC): rabbit anti-CART; Red (CY3): goat anti-leptin; Purple (false coloured, CY5): rat anti-NPY Rat brain section – confocal microscopy QUADRUPLE Labelling
- 39. Some useful websites • http://www.uchsc.edu/rmtsc/protocol/index.html • http://www.jacksonimmuno.com/ • http://www.invitrogen.com/site/us/en/home/brands/Molecular-Probes.html • http://www.biocompare.com/ProductCategories/2045/Antibodies.html • http://www.cytochemistry.net/Cytochem.htm • http://www.sigmaaldrich.com/life-science/cell-biology/antibodies.html