
Recommandé
Contenu connexe
Tendances
Tendances (20)
En vedette
En vedette (20)
Similaire à 2 md2016 annotation
Similaire à 2 md2016 annotation (20)
Dernier
Dernier (20)
2 md2016 annotation
- 2. how?
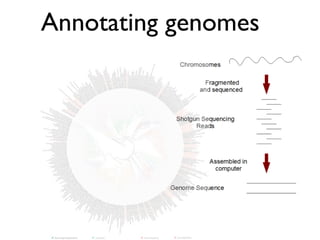
- 4. Genome Sequencing Step by step… • Library construction and sequencing • Base-calling: Quality Control • Assembly (repeat as necessary) • Annotation (repeat as necessary) • Publish!
- 5. Genomes vary greatly in size Organism Genome size (Mb) Nanoarchaeum equitans 0.49 Mycoplasma genitalium 0.58 Escherichia coli 4.68 Methanosarcina acetivorans 5.75 Saccharomyces cerevisiae 12 Caenorhabditis elegans 90 Drosophila melanogaster 180 Zea mays 2500 Homo sapiens 3500 Hordeum (barley) 5100
- 6. Bacterial/archaeal genomes range from 0.5Mb-> 13Mb and from ~500 genes to > 10,000 genes
- 7. Genome Size Variability • Extent of gene duplication • Repetitive DNA • Gene size – number and length of introns • Space between genes (% coding) – regulatory regions – heterochromatin
- 8. 8
- 9. How do genes get names? How to find genes in genomes Problems and strategies in genome annotation Databases that are useful for annotation How are genes related to other genes? RAST
- 10. How do you find the genes?
- 11. 11 How does a gene get a name?
- 12. Genome fragment of Nitrosocaldus yellowstonii How does a gene get a name? ORF = CDS = gene ?http://www.genenames.org
- 13. Automated Annotation Pipelines/Servers • Provide fast analysis of genomic sequences o gene identification & function prediction • Used to rely on information in public databases (beware!) • Now often based on re-analysis of published genomes • Rely on “curated” reference genomes like Prokka
- 14. Box 2 | Gene prediction versus gene annotation Although the terms ‘gene prediction’ and ‘gene annotation’ are often used as if they are synonyms, they are not. With a few exceptions, gene predictors find the single most likely coding sequence (CDS) of a gene and do not report untranslated regions (UTRs) or alternatively spliced variants. Gene prediction is therefore a somewhat misleading term. A more accurate description might be ‘canonical CDS prediction’. Nature Reviews | Genetics 229,500 229,000 228,500 228,000 227,500 226,500227,000 bp 5′UTR 3′UTR Gene annotation resulting from synthesizing all available evidence (two alternative splice forms) Protein evidence (BLASTX) mRNA or EST evidence (Exonerate) Gene prediction (SNAP) Start codon Stop codon More types of data help annotation
- 15. Nature Reviews | Genetics Post process gene predictions to add UTRs and alternatively spliced transcripts based on evidence Consensus- based chooser Consensus- based chooser Run battery of ab initio gene predictors Align ESTs, proteins and RNA-seq data to genome Run battery gene predictors in evidence-driven mode Run single ab initio gene predictor Best consensus CDS model for each gene Best consensus mRNA model(s) for each gene mRNA model(s) for each gene most consistent with evidence Most likely CDS model for each gene Optional manual curation using genome browser Manually curated gene models Increasing accuracy Consensus- based chooser Evidence- based chooser Best consensus CDS model for each gene Option 2: predict and choose Option 3: full-scale annotation pipelines Option 1: predict nreasintimeaneort Increasinguseofevidence Figure 2 | Three basic approaches to genome annotation and some common variations. Approaches are compared on the basis of relative time, effort and the degree to which they rely on external evidence, as opposed to
- 16. Bacteria/ Archaea Eucaryotes Genome Organization in Bacteria/Archaea vs. Eucaryotes Gene identification in Bacteria and Archaea is (mostly) easier than in Eucaryotes gene 1 gene 2 gene 3
- 17. 17 ugh to ensure acceptance of 9, 10]. There has also been a neration techniques such as g experimental methods n of a protein’s role and . These annotations would se they are based on actual than homology. Currently idence tags stating how the , however, they are often s. Including evidence quali- dea of the reliability of the concept of assigning a level is not novel, but is seldom ome of the current steps for otation and offers a guide to oblems that are encountered tion. It goes on to identify ce genomes and why choos- not always the best option. of the public sequence data- st possible next steps toward rehensive annotation with errors. erial genomes Figure 1: A generic process for bacterial genome Richardson and Watson atMhttp://bib.oxfordjournals.org/Downloadedfrom Steps in Genome Annotation
- 18. Identification of protein-coding regions Intrinsic evidence • Absence of stop codons (TAA,TGA,TAG) • Sufficient open-reading frame (ORF) length (~100 a.a.) • Presence of start codon (ATG, GTG, TTG) • Minimize gene overlap • presence of other sequence motifs (TATA, RBS, splice sites, polyA) Extrinsic evidence • Similarity to “known” genes from other organisms (HOMOLOGY) • Expression data (mRNA sequencing, proteomics) • Predicted sequence analysis (e.g., protein structure modeling)
- 20. 20 What are several ways that could explain sequence similarity between molecular sequences? What are potential pitfalls with assigning homology? How do we generally assign homology?
- 22. the Mycoplasma genitalium genome1 (Fig. 1). Where two groups’ descriptions are completely incompatible, at least one must be in error. In my analysis, there is no penalty sions – a likely occurrence because all relied on simil methods and data. This evaluation also ignores minor d agreements in annotation, and disparities in degree specificity (possibly indicating problematic overpredicti of function4 ). Therefore, the true error rate must greater than these figures indicate. There are several possible reasons why the function analyses have mistakes, as described at greater length els where5–8 . For example, it may be that the similar between the genomic query and database sequence insufficient to reliably detect homology, an issue solvab by appropriate use of modern and accurate sequence com parison procedures9,10 . A more difficult problem is accura inference of function from homology. Typical databa searching methods are valuable for finding evolutionar related proteins, but if there are only about 1000 maj superfamilies in nature11,12 , then most homologs mu have different molecular and cellular functions. The annotation problem escalates dramatically beyo the single genome, for genes with incorrect functions a entered into public databases8 . Subsequent search against these databases then cause errors to propagate future functional assignments. The procedure need cyc only a few times without corrections before the resourc that made computational function determination possib – the annotation databases – are so polluted as to almost useless. To prevent errors from spreading out control, database curation by the scientific commun will be essential4,13 . To ensure that databases are kept usable, the intent o gene annotation should be clear: does it indicate homolo ortholog, and/or functional equivalence? Fortunately, som databases already incorporate this information explici (e.g. Ref. 14). Errors will, of course, still creep in. To he FIGURE 1. Comparison of annotations Three dots represent (left to right) Frasier et al.1 , Koonin et al.2 and Ouzounis et al.3 annotations for each of the 468 M. genitalium genes. (Tentative cases 001 051 101 151 201 251 301 351 401 451 M. genitalium Black circle = no annotation 468 genes Colored circle = different Blue circle = same annotation TIG April 1999, volume 15, No. 4 13 atory of Molecular Biology, Hills Road, UK. M. Levitt, C. Chothia, B. Al-Lazikani provided stimulating discussion. No. groups No. Annotations per group Total No. annotating gene genes annotations conflicts Frasier Koonin Ouzounis et al.1 et al.2 et al.3 0 33 – – – – N/A 1b 95 14 15 66 95 N/A 2 318 279 317 40 636 45 3 22 22 22 22 66 10 Sum (2+3) 340 301 339 62 702 55 Summary of annotations made by each group (Fig. 1), minimal number of conflicting annotations (s the resulting minimal fraction of annotations that are erroneous. a Frasier et al.1 data from http://www.tigr.org/tdb/mdb/mgdb/mgdb.html. Koonin et al.2 data from ht nlm.nih.gov/Complete_Genomes/Mgen. Ouzounis et al.3 data from http://www.embl-heidelberg mycogen.new.html. Instances where Ouzounis et al.3 reported SWISS-PROT annotation of the same gene w avoid duplication with Frasier et al.1 entries. However, even if all of these 300 annotations are included annotation error rate drops only to 6%. All annotations were collected in 1996, shortly after the genom b No comparative analysis is possible when only one group made an annotation. al. (1995) The minimal gene complement of Mycoplasma nce 270, 397–403 l. (1996) Sequencing and analysis of bacterial genomes. 4–416 al. (1996) Novelties from the complete genome of Mycoplasma Microbiol. 20, 898–900 (1998) Protein annotation: detective work for function prediction. , 248–250 nd Koonin, E.V. (1998) Sources of systematic error in functional nomes: domain rearrangement, non-orthologous gene nd operon disruption. In Silico Biol. 1, 7 Zhang, X. (1997) The challenges of genome sequence annotation or he details’. Nat. Biotechnol. 15, 1222–1223 998) Predicting function: from genes to genomes and back. , 707–725 roch, A. (1996) Go hunting in sequence databases but watch out for s Genet. 12, 425–427 al. (1998) Assessing sequence hods with reliable structurally identified distant evolutionary oc. Natl. Acad. Sci. U. S. A. al. (1994) Issues in searching molecular sequence databases. 19–129 11 Chothia, C. (1992) Proteins. One thousand families for the molecular biologist. Nature 357, 543–544 12 Brenner, S.E. et al. (1997) Population statistics of protein structures: lessons from structural classifications. Curr. Opin. Struct. Biol. 7, 369–376 13 Smith, T.F. (1998) Functional genomics – bioinformatics is ready for the challenge. Trends Genet. 14, 291–329 14 Tatusov, R.L. et al. (1997) A genomic perspective on protein families. Science 278, 631–637
- 23. COMMENTErrors in genome annotation FIGURE 2. Example annotations and analysis (a) Consistent annotations. Annotations were generally considered consistent for this analysis if either the function or the gene name match (e.g. mg463; mg010). An exception is when one group uses a gene name and another specifically notes that the current gene is a paralog and not identical (consider mg010). Where the descriptions from different groups were compatible, but of different levels of specificity, this was considered a correct assignment (e.g. mg225). The difficulty of reconciling pairs of descriptions to determine whether they reflect compatible functions makes this analysis imprecise. Generally, the approach here is generous and should err on the side of detecting too few errors; it is usually more permissive than Ref. 5. mg463: Frasier et al.1 and Koonin et al.2 describe different aspects of function, but give the same gene name. The Ouzounis et al.3 description is compatible with that from Koonin et al.2 , but less specific. All three annotations are considered correct for this analysis. mg010: Frasier et al.1 and Ouzounis et al.3 agree that this is a DNA primase. Koonin et al.2 use a different gene name and explicitly state that this is a truncated protein. Because of the common functional descriptions, all three are considered correct. However, if Koonin et al.2 had been more explicit in indicating a functional difference, then their annotation would have been marked as conflicting. (Note that mg250 is also annotated as a DNA primase by all three groups.) mg225: the Ouzounis et al.3 annotation of histidine permease is more specific than the Koonin et al.2 description of amino acid permease. It may be that histidine permease is an (incorrect) overprediction of function, or it could be correct. The two annotations are considered consistent, and the decision of Frasier et al.1 not to provide a function is not penalized. (b) Inconsistent annotations. mg302: lack of a functional assignment from Frasier et al.1 is not penalized. The Koonin et al.2 and Ouzounis et al.3 annotations are wholly inconsistent. This leads to a conflict and a minimum error rate of 50%. Note that the assessment (a) mg463 Frasier et al. High level kasgamycin resistance (ksgA) Koonin et al. rRNA (adenosine-N6, N6-)-dimethyltransferase (ksgA) Ouzounis et al. Dimethyladenosine transfe [sic] mg010 Frasier et al. DNA primase (dnaE) Koonin et al. DNA primase (truncated version) (DnaGp) Ouzounis et al. DNA primase (EC 2.7.7.-) mg225 Frasier et al. Hypothetical protein Koonin et al. Amino acid permease Ouzounis et al. Histidine permease (b) mg302 Frasier et al. No database match Koonin et al. (Glycerol-3-phosphate?) permease Ouzounis et al. Mitochondrial 60S ribosomal protein L2 mg448 Frasier et al. Pilin repressor (pilB) Koonin et al. Putative chaperone-like protein Ouzounis et al. PilB protein mg085 Frasier et al. Hydroxymethylglutaryl-CoA reductase (NADPH) Koonin et al. ATP(GTP?)-utilizing enzyme Ouzounis et al. NADH-ubiquinone oxidoredu [sic] Two kinds of problems insufficient similarity to assume homology inference of function from homology
- 24. 24 Table 1 Statistics for different annotations for H. utahensis genome along with the extended annotations. For orphan and functional genes genes and the percentage relative to the total number of annotated genes Annotation features NCBI AAMG RAST Extend Original Complemented by annotation of function from AAMG and RAST Original Complemented by annotation of function from NCBI and RAST Original Complemented by annotation of function from NCBI and AAMG EA CDS 2998 2998 3040 3040 3041 3041 2980 rRNA 4 4 3 3 3 3 4 tRNA 45 45 45 45 45 45 45 ncRNA 1 1 0 0 0 0 0 frameshift/Pseudo 0 0 0 0 0 0 0 Total 3048 3048 3088 3088 3089 3089 3029 Orphan genes 1014 (33.27 %) 777 (25.49 %) 885 (28.66 %) 837 (27.10 %) 1203 (38.94 %) 819 (26.51 %) 672 (22 Functional genes 2034 (66.73 %) 2271 (74.51 %) 2203 (71.34 %) 2251 (72.90 %) 1886 (61.06 %) 2270 (73.49 %) 2357 (7 Another issue with annotation Not all proteins have homologs in another genome — check out Giardia
- 25. Reflecting annotation uncertainty in gene names • “Domain”-containing protein Predicted protein contains a region similar to a recognized protein domain or fold – ankyrin-repeat domain containing protein • Conserved hypothetical protein Predicted protein is homologous to predicted proteins in at least one other (distinct!) organism • Hypothetical protein Nothing is known about the predicted protein (no known homologs) Avoid “-like” as homology is a yes/no
- 26. Dangers of Serial Annotation • Function is generally “inferred” from homology • Poor annotations are propagated in the public sequence databases (GenBank) - think the Telephone Game • Failure to examine functional assignation leads to deterioration of data and errors • Manual curation is needed to validate annotation and add valuable information • Particularly important for representatives of new lineages – often homologous genes in new lineages are very different from those in other organisms – need good annotation of “anchor” genomes for subsequent sequencing
- 27. 27 Leave databases better than you found ‘em Keep your messy thoughts outta databases.
- 28. 28 How does Prokka work?
- 29. Does Loki have actin?
- 30. 30 0.4 LCGC14AMP_05736710 Crenactin LCGC14AMP and Lokiarchaeum (4/1) Actin and related sequences Arp2 LCGC14AMP (5) Arp1 LCGC14AMP and Lokiarchaeum (11/1) LCGC14AMP/Lokiarchaeum (11/2) LCGC14AMP (2) Arp3 LCGC14AMP (2) LCGC14AMP_06532160 100 100 51 83 100 100 96 100 100 100 100 a c b Nitrosopumilus maritimus SCM1 0 LCGC14AMP and Lokiarchaeum (5/1) RESEARCH ARTICLE What if you found eukaryotic genes in an archaeon? 30
- 32. 32 How do genes evolve? Speciation —> Diversification Gene duplication —> Diversification
- 33. Almost half of the genes in any genome are in gene families 33 and are deleted from the genome. The rate of duplication that gives rise to stably maintained genes is the birth rate multiplied by the retention rate, which is expected to fluctuate with gene function, among other things. Duplicated genes are often referred to as paralogous genes, which form gene families. Several authors have tabulated the distribution of gene family size for a few completely sequenced genomes [11,12] and this varies substantially among species and gene families [13]; for instance, the biggest gene family in D. melanogaster is the Table 1. Prevalence of gene duplication in all three domains of lifea Total number of genes Number of duplicate genes (% of duplicate genes) Refs Bacteria Mycoplasma pneumoniae 677 298 (44) [65] Helicobacter pylori 1590 266 (17) [66] Haemophilus influenzae 1709 284 (17) [67] Archaea Archaeoglobus fulgidus 2436 719 (30) [68] Eukarya Saccharomyces cerevisiae 6241 1858 (30) [67] Caenorhabditis elegans 18 424 8971 (49) [67] Drosophila melanogaster 13 601 5536 (41) [67] Arabidopsis thaliana 25 498 16 574 (65) [69] Homo sapiens 40 580b 15 343 (38) [11] a Use of different computational methods or criteria results in slightly different estimates of the number of duplicated genes [12]. b The most recent estimate is ,30 000 [61].
- 34. Paralogous Gene Families - many genes in the genome are present in “families” and each gene in a gene family shares a common ancestry (homologs) - gene families arise from duplication and subsequent diversification by various mechanisms how are these copies different from alleles? full duplicated dead new ancestral Possible fates of duplicated genes: sub
- 35. Evolutionary fates of duplicated genes pseudogenization - non-functional when accumlates a stop codon. this gene is eventually lost from genome. but young pseudogenes would still recognizable as a homolog. why? 35 conservation of function - extra copy could provide greater amounts of protein. why?
- 36. 36 subfunctionalization - extra copy could have a new function (or a sub-function). why? – most proteins have > 1 function (could be expressed differently in different parts of cell/tissue or at different times) – if greater amounts of a protein not advantageous, extra copy would be selected against unless… – subfunctionalization - both copies adopt some functions of parent gene (moonlighting functions) – sometimes this can be differential gene expression in different tissues
- 37. 37 Neofunctionalization - extra copy could have a novel function. why? –often a related function (not entirely new) –opsin gene family is a good example –this could require a lot of mutations in new gene copy
- 38. Orthologs and paralogs a A*b* c BC* Ancestral gene Duplication to give 2 copies = paralogs on the same genome orthologousorthologous paralogous A*C*b* A mixture of orthologs and paralogs sampled potential problem
- 39. Orthologs: Homologs inherited after speciation. Gene phylogeny may match organismal phylogeny. Paralogs: Homologs produced by gene duplication. Multiple homologs in a given species or evidence that gene duplication involved through phylogenetic analysis and lack of match to organismal phylogeny Gene phylogeny does not match organismal phylogeny in a tree where most genes do match organismal phylogeny well.
- 40. Using phylogeny to check for paralogs (or orthologs) –multiple copies of a gene in a genome –look at which clades contain the paralogs –duplication events can occur > once –can be paralog loss/gain 40
- 41. 41 actin ARPs crenactin 41 0.4 LCGC14AMP_05736710 Crenactin LCGC14AMP and Lokiarchaeum (4/1) Actin and related sequences Arp2 LCGC14AMP (5) Arp1 LCGC14AMP and Lokiarchaeum (11/1) LCGC14AMP/Lokiarchaeum (11/2) LCGC14AMP (2) Arp3 LCGC14AMP (2) LCGC14AMP_06532160 100 100 51 83 100 100 96 100 100 100 100 a c b Lokiar Eur Lokiarch_12 Arf-family Lok Lokiarc 170290521 C Lokiarch_31930 Lokiarchaeum Lok Lokiarch Sar1-fam Lokiarch 3154254 Lokiarchaeum (3) Rab-family (7 5 Lokiarchaeum (4) Lokiarchae Lokiarch_45420 Lokiarc 51 79 100 99 100 71 87 99 84 96 97 89 100 95 82 93 69 100 68 61 82 99 97 Arabidopsis thaliana Thalassiosira pseudonana Methanopyrus kandleri AV19 Pyrobaculum aerophilum IM2 Aciduliprofundum boonei T469 Korarchaeum cryptofilum OPF8 Caldiarchaeum subterraneum Myxococcus xanthus DK1622 Nitrosopumilus maritimus SCM1 0 3 1 2 2 4 31 113 4 LCGC14AMP and Lokiarchaeum (5/1) RESEARCH ARTICLE Actins are part of a gene family ARP = actin related protein
- 42. 42 from sequence-based homology searches (Fig. 1). Despite this variance, two features are preserved between prokaryotic and eukaryotic actins. The first common feature in multistrand filament architectures. This maintenance of contacts within a strand suggests that the primordial actin filament was single-stranded. In PNAS, Braun et al. packing (6, 7). L crographs (EMs) indicated a struc either single- or (6). Now, Braun in an 18-Å cry crenactin can for in vitro. In determinin sents a record o actin filament, functions must appears to inte proteins, the arca as a cell shape-d ment has some actin homologs M bulin homolog F a dedicated cell s idence from bact filaments have a sequences and f evolved to becom tion (1). Conseq why crenactin fo ment may be tha mal for its role Fig. 1. Relatedness of actins. The structures of actin protofilaments (2, 9–15) are shown below a maximum- likelihood phylogenetic tree of the actin protein sequences. The structures are aligned via the central protomer, Author contributions: U.G Actins are part of a larger gene family
- 43. 43 actin crenactin MreB 43 0.4 LCGC14AMP_05736710 Crenactin LCGC14AMP and Lokiarchaeum (4/1) Actin and related sequences Arp2 LCGC14AMP (5) Arp1 LCGC14AMP and Lokiarchaeum (11/1) LCGC14AMP/Lokiarchaeum (11/2) LCGC14AMP (2) Arp3 LCGC14AMP (2) LCGC14AMP_06532160 100 100 51 83 100 100 96 100 100 100 100 a c b 68 Pyrobaculum aerophilum IM2 Aciduliprofundum boonei T469 Korarchaeum cryptofilum OPF8 Caldiarchaeum subterraneum Myxococcus xanthus DK1622 Nitrosopumilus maritimus SCM1 0 3 1 2 2 4 LCGC14AMP and Lokiarchaeum (5/1) RESEARCH ARTICLE Actins are part of a gene family Actin and Arp 2/3 required for motility Arps = actin related proteins (and are not actin)
- 44. 44 centerofthetree)but,exceptincarefullycalibratedcases,thisrelationshipis not defined and probably varies between different parts of the tree. defining subgroups by the deepest strongly supported node. Modified, with permission, from Ref. [3]. Sc ARP4 Sp P23A10.08 Sp C23D3.09 Ce ZK616.4 Dm CG6546 Hs BAF53b Mm BAF53a Hs BAF53a Sc ARP7 Sc ARP9 Sp C1071.06 Ce F42C5.9 At 8843903 Sc ARP8Sp C664.02Dm CG7846 Mm 12857259 Hs 104344709 Dm CG12235 Hs 'ARP11'Ce C49H3.8 M m 'ARP11' Sp C56F2 Sc ARP10 HsARP5 Dm CG7940 ScARP5 ScARP6 CeARP6 Dmactin13E GgARPX Mm 'Actlike7b' Hs'Actlike7b' AtARP3 ScARP3 Dm actin66b Hs ARP3Mm 12835802 At ARP2 Sc ARP2 Dd ARP2 Dm ARP14D ScARP1NcARP1 DmARP87C Nc Ro7 At12321978 Os13486900 SpBC365.10 At6091748 SpCC550.12 Mm12842577 HsARPX At11276982Hs11137605 Mm12838437 Hs10178893 Mm 'Actlike7a' Hs'Actlike7a' Hs ARP3b Sp ARP3 D d AR P3 Ac AR P3 CeY71F9AL.16 NcARP3 Sp ARP2 Ac ARP2 Gg ARP2 Ce K07C5.1 Hs ARP2 M m 12840619 Hs 13383265 M m 12840134 CeY53F4B.22 AnARP1 Sp ARP1 HsARP1bMmARP1b MmARP1a HsARP1a TgActin GlActin DmARP53d PfActin SpActinScActin AtActin Dd actin Metazoan actins Conventional actins ARP1 Dynein motility (dynactin complex) ARP3 Actin polymerization (ARP 2/3 complex) ARP6 Nuclear? ARP5 Chromatin remodeling ARP4 Chromatin remodeling ARP8 Chromatin remodeling ARP10? Dynein motility (dynactin complex) Confidence estimates: ARP2 Actin polymerization (ARP2/3 complex) At 18394608
- 45. 45 actin arps crenactin 45 0.4 LCGC14AMP_05736710 Crenactin LCGC14AMP and Lokiarchaeum (4/1) Actin and related sequences Arp2 LCGC14AMP (5) Arp1 LCGC14AMP and Lokiarchaeum (11/1) LCGC14AMP/Lokiarchaeum (11/2) LCGC14AMP (2) Arp3 LCGC14AMP (2) LCGC14AMP_06532160 100 100 51 83 100 100 96 100 100 100 100 a c b 68 61 Caldiarchaeum subterraneum Myxococcus xanthus DK1622 Nitrosopumilus maritimus SCM1 0 3 1 LCGC14AMP and Lokiarchaeum (5/1) RESEARCH ARTICLE How confident are you in the functions of the Loki actin homologs based on this tree?
- 46. Some types of Protein Databases Database Advantages Problems nr (Translated GenBank sequences) • Everybody can submit data • Many errors, because there is no manual inspection • no additional information links • redundant UniProt (Trembl) • non-redundant dataset derived from GenBank, DDBJ and Embl • Links to additional information • GO term annotations Many errors, because there is no manual inspection RefSeq • mostly fully sequenced organisms • data submitted by genome projects • some entries are reviewed • less links to other databases • Not so many sequences as in nr and Trembl UniProt (SwissProt) • All entries reviewed • Links to additional information • Not so many sequences as in nr and Trembl and RefSeq Annika Joecker Max-Planck Institute for Plant Breeding Research
- 47. Annika Joecker Max-Planck Institute for Plant Breeding Research Sources of Information Many types of databases are used for genome annotation
- 48. 09/29/10 Plant Computational Biology UniProt – Cross-references GO Interpro Pfam
- 49. CDD – Conserved Domain Database • Contains protein domain models imported from Pfam, SMART, COG (clusters of orthologous genes), KOG (euk COGs) • Curated and provided at NCBI • Search tool: RPSBlast • 27036 PSSMs (Position specific scoring matrices) (Dec 2008) –Count amino acids at each position in multiple alignment –Compute percentage –Compute log ratio Annika Joecker Max-Planck Institute for Plant Breeding Research
- 50. Protein Domain Search: InterPro • Database of protein families, domains and functional sites • Hosted at the European Bioinformatics Institute (EBI) • Consortium of member databases (PROSITE, Pfam, Prints, ProDom, SMART and TIGRFAMs, Superfamily, Panther) • Tool for searching: InterProScan • http://www.ebi.ac.uk/Tools/InterProScan/ Annika Joecker Max-Planck Institute for Plant Breeding Research
- 51. KEGG – Kyoto Encyclopedia of Genes and Genomes • Comprehensive database of biological information: –KEGG GENES: genes and proteins –KEGG LIGAND: endogenous & exogenous chemical building blocks –KEGG PATHWAY: biochemical pathways –KEGG BRITE: KEGG-based ontology • Web and stand-alone based tools
- 52. • A way to capture biological knowledge in a written and computable form The Gene Ontology • A set of concepts and their relationships to each other arranged as a hierarchy www.ebi.ac.uk/QuickGO Less specific concepts More specific concepts
- 53. Ontologies: The Scope of GO 1. Molecular Function e.g. protein kinase activity 2. Biological Process e.g. cell cycle 3. Cellular Component e.g. mitochondrion GO terms aim to describe the ‘normal’ functions/ processes/locations that gene products are involved in NO: pathological processes, experimental conditions or temporal information
- 54. Anatomy of a GO term 7/31/14 a. b. c.
- 55. Microbes Online www.microbesonline.org • Excellent resource for microbial genome data • Precomputed ortholog/ paralog searches • Aligned protein sequences for phylogenetic analysis • Pathway-based organization of data
- 56. Microbes Online
- 57. Microbes Online
- 58. RAST and SEED viewer
- 59. 59
- 60. 60
- 61. 61 darker colors = higher % similarity
- 63. How to annotate metagenomic data? all the ways we’ve discussed before (think homologs....)
- 64. phylogenetic “binning” 40.2 Methods for the Phylogenetic Binning of Metagenome Sequence Samples root Gammaproteobacteria Proteobacteria Deltaproteobacteria Epsilonproteobacteria Betaproteobacteria Alphaproteobacteria Bacteroidia Bacteroidetes Firmicutes Bacilli Clostridia Archaea Euryarchaeota Thermoprotein Methanomicrobia Bacteria Actinobacteria (class) Actinobacteria Cyanobacteria Spirochaetes Actinobacteria (class) Actinobacteria root Archaea Bacilli Euryarchaeota Methanobacteria Bacteria Firmicutes Clostridia (B) (A) Figure 40.1 Comparison of composition of public database microbial community analyzed sequencing. (A) Taxonomic co finished genomes present in Ge May 2009: The large bias towa Gammaproteobacteria is caused by 164 genome sequences of E strains. (B) Taxonomic compos populations in the human gut e genbank metagenomic data from human gut 1. homology-based assignment of reads (e.g., BLAST) 2. compositionally-based assignment (e.g, %G+C, or nucleotide frequencies - stretches of 2-9 nts) assigning genomic data to different groups of organisms