Pancitopenia. Diagnostico diferencial
•Descargar como PPTX, PDF•
7 recomendaciones•6,885 vistas

Pancitopenia. Diagnostico diferencial

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Pancitopenia. Diagnostico diferencial
Similar a Pancitopenia. Diagnostico diferencial (20)
Más de eddynoy velasquez
Más de eddynoy velasquez (20)
Último
Último (20)
Pancitopenia. Diagnostico diferencial
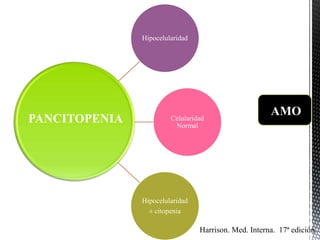
- 1. Hipocelularidad Celularidad Normal Hipocelularidad ± citopenia PANCITOPENIA Harrison. Med. Interna. 17ª edición. AMO
- 2. Celularidad normal Línea Blanca:. Leucopenia marcada. Línea Roja: Normociticos, Hipocromicos, con disminución en el numero Línea Plaquetaria: Plaquetopenia
- 3. ENFERMEDADES PRIMARIAS DE LA MÉDULA ÓSEA - Síndromes mielodisplásicos - Hemoglobinuria paroxística nocturna -Mielofibrosis Algunas leucemias a leucémicas -Mieloptisis -Linfoma de la médula ósea - Tricoleucemia SECUNDARIAS A ENFERMEDADES GENERALES - LES - Hiperesplenismo - Déficit de B12 ó de folato - Infecciones fulminantes - Alcohol - Brucelosis - Sarcoidosis - Tuberculosis - Leishmaniosis Harrison. Med. Interna. 17ª edición.
- 6. ENFERMEDADES PRIMARIAS DE LA MÉDULA ÓSEA - Síndromes mielodisplásicos - Hemoglobinuria paroxística nocturna -Mielofibrosis Algunas leucemias a leucémicas -Mieloptisis -Linfoma de la médula ósea - Tricoleucemia Harrison. Med. Interna. 17ª edición.
- 7. La mieloptisis se define como la infiltración de la MO por células no hematopoyéticas, lo que resulta en diferentes grados de anemia, trombocitopenia y neutropenia. Harrison. Med. Interna. 17ª edición.
- 8. La anemia aplásica es una pancitopenia vinculada a hipocelularidad de la médula ósea. Se debe a lesión grave el compartimiento de células hematopoyéticas Puede aparecer subitamente ó tener un comienzo gradual Las hemorragias suelen ser el primer síntoma (sangrado fácil con los roces, hemorragias gingivales, epistaxis, períodos menstruales abundantes y petequieas) Es habitual encontrar petequias y equimosis Puede haber hemorragias retinianas Palidez de piel y mucosas No es común encontrar adenopatías ó esplenomegalia. Harrison. Med. Interna. 17ª edición. El FSP muestra eritrocitos grandes con plaquetas y granulocitos escasos
- 9. Los SMD constituyen un grupo heterogéneo de desórdenes hematológicos clonales adquiridos, que afectan la célula madre hemocitopoyética y se caracterizan morfológica y clínicamente por: hematopoyesis ineficaz, Progresiva citopenia periférica, Displasia en uno o más linajes celulares, Médula ósea (MO) hipercelular y displásica con porcentaje variable de blastos Tendencia evolutiva a leucemia aguda (LA) La presencia de células blásticas en cantidades variables no supera el 20% del total de las células de MO. Se debe sospechar siempre un SMD en aquellos pacientes adultos, generalmente mayores de 50 años, que presentan una alteración en sangre periférica persistente o con un mínimo de evolución de 6 meses que no tenga una clara explicación clínica. Síndromes mielodisplásicos
- 10. En la Mielofibrosis hay una expansión clonal de la célula madre hematopoyética que se acompaña de una proliferación reactiva no-clonal de los fibroblastos y fibrosis de la médula ósea. A medida que la médula ósea se hace fibrótica la hematopoyesis no se puede mantener y ocurre una hematopoyesis extramedular (metaplasia mieloide) en el hígado y el bazo. En mas del 50% casos en el AMO, no se obtiene muestra. Harrison. Med. Interna. 17ª edición.
- 11. La Tricoleucemia o Leucemia de Células Peludas (LCP), es una enfermedad linfoproliferativa crónica, caracterizada por esplenomegalia, pancitopenia e infecciones recurrentes.
- 12. La HPN es una anemia hemolítica crónica adquirida que se caracteriza por hemólisis intravascular persistente sujeta a exacerbaciones recidivantes. Además de la hemólisis suele haber pancitopenia La mayoria de pacientes son adultos jóvenes Ocurre hematuria macroscopica La médula ósea suele ser celular con hiperplasia eritroide de notable a masiva con características diseritropoyéticas Harrison. Med. Interna. 17ª edición.
- 15. SECUNDARIAS A ENFERMEDADES GENERALES - Hiperesplenismo - Déficit de B12 ó de folato - Infecciones fulminantes - Alcohol - Brucelosis - Sarcoidosis - Tuberculosis - Leishmaniosis - LES Harrison. Med. Interna. 17ª edición.
- 16. Harrison. Med. Interna. 17ª edición. Este trastorno se caracteriza por : • Esplenomegalia • Aumento células inmaduras en sangre • Médula ósea normal ó hiperplásica
- 17. Harrison. Med. Interna. 17ª edición. La causa suele ser una deficiencia de cobalamina ó folato. Manifestaciones clínicas • Anorexia, Pérdida de peso, Glositis, Queilosis Laboratorio: • Volumen corpuscular medio (VCM) elevado • Deficiencia de folato ó cobalamina Sangre periférica • Presencia de macrocitos ovalados • VCM mayor de 100 fl Médula ósea La médula ósea es hipercelular con acumulación de células primitivas.
- 18. Harrison. Med. Interna. 17ª edición. Relacionado con cirrosis hepática.
- 19. • Enfermedad inflamatoria caracterizada por la presencia de granulomas no caseosos • Suele afectar múltiples órganos y sistemas, y para confirmar el diagnóstico específico se necesita que haya afección de dos ó más de ellos.
- 20. Manifestaciones: • Fiebre prolongada • Caquexia • Hepatoesplenomegalia • Pancitopenia • Hipoalbuminemia • Ulceras que remiten de manera espontanea
- 22. PATRON: PERIFERICO VALOR: 1:256 El patrón periférico se caracteriza por tinción regular alrededor del núcleo; el centro de este patrón muestra menos tinción. La placa de la cromatina se tiñe de forma delineada o compacta.
- 25. Enfermedad autoinmune crónica en la que los órganos, tejidos y células se dañan por la adherencia de diversos autoanticuerpos y complejos autoinmunitarios. Harris: Kelley's Textbook of Rheumatology, 7th ed.,
- 26. Afectación multisistemica. Distribución universal. Mujeres en edad fértil. Pico de incidencia 15 y 40 años Mujeres/hombres 10/1 Principios de Reumatologia Klippel 12a edicion Cap 17
- 27. La prevalencia del lupus va de aproximadamente 40 casos por cada 100.000 personas entre los europeos del norte. Predomina en la raza negra con prevalencia de 200 por cada 100.000 personas. En los Estados Unidos, el número de pacientes con lupus excede las 250.000. La esperanza de vida de estos pacientes ha mejorado de una tasa de supervivencia de aproximadamente 4 años (1950) a una tasa de supervivencia a 15 años. Systemic Lupus Erythematosus. N Eng J Med 2008;358:929-29
- 28. Sistémicas. (95%) Musculoesqueléticas.(95%) Hematológicas(85%) Cutáneas.(80%) Cardiopulmonares.(60%) Neuropsiquiatricas.(60%) Renales.(50%) Manual CTO 7a edicion
- 30. Bordes eritematosos, hiperpigmentados, escamosos y ligeramente elevados con centros atróficos y despigmentados donde existe destrucción permanente de los apéndices dérmicos. Las lesiones llegan a causar desfiguración, en la cara y el cuero cabelludo.
- 33. Harris: Kelley's Textbook of Rheumatology, 7th ed.,
- 34. Reducir las exacerbaciones agudas Suprimir los síntomas lo más pronto posible Evitar el daño orgánico OBJETIVOS DELTRATAMIENTO
- 35. Indicaciones Se inicia con corticoides sistémicos a dosis altas Paciente tiene alguna manifestación grave de riesgo vital Antes de administrarlos descartar la presencia de infección
- 36. Administración de glucocorticoides 0.5 a 2 m g / k g / día por vía oral o 1 000 mg de metilprednisolona IV C/dia x tres días. 0.5 a 1 mg/kg/día de prednisona x 4 a 6 semanas se reducen de manera gradual. Dosis de mantenimiento de 5 a 10 mg/día de prednisona o su equivalente o entre 10 y 20 mg cada tercer día. LES POTENCIALMENTE LETAL: VARIEDADES PROLIFERATIVAS DE NEFRITIS POR LUPUS
- 37. Ciclofosfamida (un alquilante) Micofenolato de mofetilo un inhibidor relativamente específico del linfocito que tiene acción sobre la monofosfatasa de inosina y por tanto en la síntesis de purina. Azatioprina (un análogo de purina y antimetabolito específico de ciclo) Tratamiento de la nefritis
- 38. Los farmacos citotoxicos estan indicados en: 1 Enfermedad grave con afectación de órgano vital que no responde a dosis elevada de Glucocorticoides. 2 Afectación de órgano vital que recurre al reducir la dosis de Glucocorticoides. 3 Toxicidad o efectos adversos de los Glucocorticoides en enfermedad activa. 4 Formas activas de glomerulonefritis proliferativa difusa Harris: Kelley's Textbook of Rheumatology, 7th ed.,
- 39. Ciclofosfamida dosis recomendada es de 500 a 750 m g / m 2 por vía intravenosa, cada mes durante tres a seis meses. Estudios europeos han sugerido que la ciclofosfamida en dosis totales de 500 mg cada dos semanas hasta seis dosis tiene la misma eficacia que las dosis más altas y duraciones más prolongadas antes recomendadas, durante un periodo de cinco a siete años.
- 40. Es grave Mortal si no se trata sobre todo en los 3 primeros años de evolución La principal causa de muerte son: Infecciones Nefropatía Afectación del sistema nervioso central
- 41. The American Journal of Medicine Volume 119, Issue 8
- 43. CASO DE MEDICINA INTERNA N° 12- 3431
- 50. DIAGNOSTICO: BIOPSIA DE PIEL DE MEJILLA DERECHA: ECCEMA CRONICO COMPATIBLE CON LUPUS ERITEMATOSO DISCOIDE