Comparison of Clinical Trial Application requirement of India, USA and Europe.
•
26 j'aime•5,665 vues

Comparison of Clinical Trial Application requirement of India, USA and Europe.

Recommandé
Recommandé
Contenu connexe
Tendances
Tendances (20)
En vedette
En vedette (12)
Similaire à Comparison of Clinical Trial Application requirement of India, USA and Europe.
Similaire à Comparison of Clinical Trial Application requirement of India, USA and Europe. (20)
Plus de Aakashdeep Raval
Plus de Aakashdeep Raval (13)
Dernier
Dernier (20)
Comparison of Clinical Trial Application requirement of India, USA and Europe.
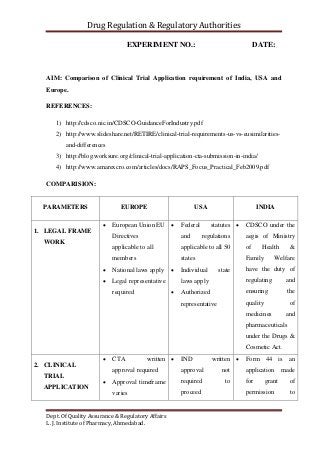
- 1. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. EXPERIMENT NO.: DATE: AIM: Comparison of Clinical Trial Application requirement of India, USA and Europe. REFERENCES: 1) http://cdsco.nic.in/CDSCO-GuidanceForIndustry.pdf 2) http://www.slideshare.net/RETIRE/clinical-trial-requirements-us-vs-eusimilarities- and-differences 3) http://blog.worksure.org/clinical-trial-application-cta-submission-in-india/ 4) http://www.amarexcro.com/articles/docs/RAPS_Focus_Practical_Feb2009.pdf COMPARISION: PARAMETERS EUROPE USA INDIA 1. LEGAL FRAME WORK European Union EU Directives applicable to all members National laws apply Legal representative required Federal statutes and regulations applicable to all 50 states Individual state laws apply Authorized representative CDSCO under the aegis of Ministry of Health & Family Welfare have the duty of regulating and ensuring the quality of medicines and pharmaceuticals under the Drugs & Cosmetic Act. 2. CLINICAL TRIAL APPLICATION CTA written approval required Approval timeframe varies IND written approval not required to proceed Form 44 is an application made for grant of permission to
- 2. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. Annual safety report only required Format CTD paper or electronic National CTA fees may apply commence CT May proceed 30- days after FDA receives IND unless notified otherwise IND annual report required Format paper or electronic, US format or CTD No fees Required import or manufacture a new drug or to undertake Clinical Trial. documents pertaining to chemical and pharmaceutical information, animal pharmacology, toxicology data and clinical pharmacology data. Investigator’s Brochure, trial protocol, case report form, informed consent form, patient information sheet, investigator’s undertaking and IEC approvals (if obtained during review process). Regulatory status of the trial in other participating countries also needs to be
- 3. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. reported. Fees for phase I application is Rs 50000 Fees for phase II and III application is 25000 3. INSTITUTIONAL REVIEW BOARD EC approval required ECs appointed or authorized by States IRB approval required IRB registration required DCGI approval IRB / EC approval required 4. FORM/S REQUIRED Statement of Investigator not required by member states Form FDA 1572 is required to be signed by the PI, if study is conducted in US and submitted to IND Form no: 44 is required for application of clinical trial. 5. RECORD RETENTION Essential Document Record includes CRF, excluding medical records: ≥ 5 years ≥ 15 years or CT discontinuation if data used to support a marketing application Record retention 2 years after marketing application is approved Record retention 2 years after last shipment and delivery of IMP if marketing application is not approved. Retention of records 3 years after marketing application is approved
- 4. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. 6. INVESTIGATION AL MEDICINAL PRODUCT REQUIREMENTS Label must comply with Annex 13 of EU Directive 2001/83/EC Language requirements varies between member states Sponsor is responsible for destruction of unused and/or returned IMP Label must be in English, except for Puerto Rico The following statement is required: “Caution: New Drug Limited by Federal (or United States) law to investigational use” Study code API and formulation Batch no. , expiry date and retest date Dosage Direction of use Manufactured by “FOR CLINICAL TRIAL USE ONlLY” 7. ADVERSE EVENT REPORTING Review and monitors the safety information of IMPs used in clinical trials conducted in their respective territories through the use of the EudraVigilance Clinical Trial module (EVCTM) Report to the sponsor all serious adverse events immediately Required to report to the Sponsor any adverse events caused by or probably caused by IMP Notify FDA and all participating investigators in a written IND safety report of: Any adverse experience associated with the use of the drug that is both serious and unexpected. Required to report unexpected fatal or life threatening Upon the discovery of any injury or death related to a clinical trial, sponsors will now be required to inform the DCGI within 24 hours. Following this reporting line, relevant clinical trial stakeholders will be required to submit individual reports for scrutiny by an independent review committee, due to be created
- 5. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. experiences ASAP, but not later than 7 days; Follow-up reports ASAP but no later than 15 days of receipt of new information by the DCGI. 8. REGULATORY COMPLIANCE covered under Article 15 of Directive 2001/20/EC CA responsible for implementing provisions for the suspension of a CT conducting inspections and verifying compliance Inspection reports may also be made available to Sponsor, EC, EMEA and other member states Must comply with Good Distribution Practices (GDPs) and Good Laboratory All clinical trials must comply with 21 CFR Parts 50, 54, 56, 58 and 312 Phase 1 IMPs are exempt from certain parts of 21 CFR Part 211, unless the clinical trial involves a marketed drug product or one that was manufactured in a Phase 2 and/or 3 study Indian GCP states that if the sponsor is a foreign company, organization or person(s) – it shall appoint a local representative or CRO to fulfill the appropriate local responsibilities as governed by the Indian regulations.
- 6. Drug Regulation & Regulatory Authorities Dept. Of Quality Assurance & Regulatory Affairs L. J. Institute of Pharmacy, Ahmedabad. Practices (GLPs) no comparable EU regulation specific to Phase 1 CGMPs (Annex 13 guideline provides flexibility dependent upon the stage of development of the product)