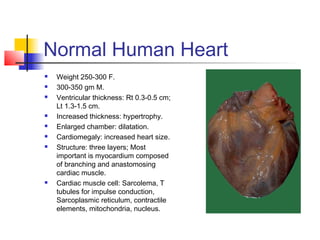
1. Normal Human Heart
Weight 250-300 F.
300-350 gm M.
Ventricular thickness: Rt 0.3-0.5 cm;
Lt 1.3-1.5 cm.
Increased thickness: hypertrophy.
Enlarged chamber: dilatation.
Cardiomegaly: increased heart size.
Structure: three layers; Most
important is myocardium composed
of branching and anastomosing
cardiac muscle.
Cardiac muscle cell: Sarcolema, T
tubules for impulse conduction,
Sarcoplasmic reticulum, contractile
elements, mitochondria, nucleus.
2. Normal Heart Structure
Mitochondria are numerous.
Sarcomere is the functional unit 1.6- 2.2 um, Z line demarcates the
sarcomere.
Cardiac muscle is more complex.
Atrial muscle cells contain granules containing atrial natriuretic peptide-
hypertension and CCF.
Intercalated discs join individual myocytes.
Gap junctions: clusters of plasma membrane channels that directly
connect the cytoplasmic components of neighboring cells, facilitate
synchronous myocyte contraction.
Abnormalities in the spatial distribution of gap junctions and their
respective proteins in IHD and myocardial disease contributes to
electromechanical dysfunction called arrhythmias.
Specialized excitatory and conducting myocytes are involved in
regulation of rate and rhythm of the heart.
AV node, bundle of his, Lt and Rt branches.
3. Blood Supply of Heart
Heart is supplied by three coronary arteries.
Rt coronary artery, Rt aortic sinus supplies Rt atrium
and ventricle, interatrial septum, SA and AV nodes,
part of Lt atrium and Lt ventricle.
Lt coronary artery, Lt aortic sinus supplies Lt ventricle
and atrium, interventricular septum, AV bundles.
Epicardial coronary arteries.
Blood supply is maximum during ventricular diastole.
Although coronaries are end arteries, collateral exist
between them which get functional if one of them is
narrowed with resulting hypoperfusion.
4. Effects of Aging on Heart
Increased Lt atrial chamber size.
Decreased Lt ventricle cavity size.
Sigmoid shape of interventricular septum.
Calcification of aortic and mitral valves.
Thickening of leaflets.
Tortuosities of coronary arteries, increased lumina, calcifications
and atherosclerotic plaques.
Increased myocardial mass.
Increased subepicardial fat.
Brown atrophy.
Lipofuscin deposition.
Basophilic degeneration.
Decreased myocytes, increased collagen.
5. Aortic Changes
Dilated ascending aorta and rightward shift
Tortuous thoracic aorta
Elastic tissue fragmentation and deposition of
collagen
Atherosclerosis
7. Cardiac Dysfunction
Failure of pump action.
Obstruction to outflow.
Regurgitation of the pumped blood.
Cardiac conduction defects.
Disruption of circulatory system.
8. Heart Failure
Heart is unable to pump blood at a rate required for tissue
metabolism.
Compensatory mechanisms are frank starling phenomenon,
hypertrophy, increased heart rate by norepinephrines, activation
of renin angiotensin aldosterone system, release of atrial
natriuretic peptide.
Most instances of heart failure are consequences of progressive
deterioration of myocardial contractile function as in IHD,
volume overload or cardiomyopathies.
Causes of heart failure are hypertension, IHD and pericarditis.
Congestive heart failure; Diminished cardiac out put and
damming back of blood in the venous system.
In CCF, the morphologic changes are often distant from heart
produced by the hypoxic and congestive effects of the failing
circulation.
9. Failure and Cardiac Hypertrophy
Initially both left and right heart failure may produce independent
signs and symptoms, but as heart is a close circuit, one sided
failure produces burden on other, ultimately leading to failure of
both units.
Causes are chronic hypertension and aortic stenosis.
Thickness of the myocardium is increased and cavity
decreases.
Regurgitation: both hypertrophy and dilatation occurs.
Total weight of the heart may be increased up to 3 times of
normal.
Hypertrophy may initially compensate for the decreased
function, but of course, the hypertrophied heart is not normal
because of deleterious structural, biochemical and molecular
alterations in the myocardium.
Sustained hypertrophy evolves into failure.
10. Right Sided Heart Failure
Usually secondary consequence.
Increased burden on right heart.
Pure right failures occurs in severe pulmonary hypertension- cor
pulmonale.
Right ventricle hypertrophied and dilated.
Findings are: 1- minimal pulmonary congestion, 2- engorged
systemic and portal veins.
Liver shows increased size and weight- congestive
hepatomegaly.
Liver lobules show red centres surrounded by pale peripheral
regions.
Hypoxia may produce centrilobular necrosis.
Sinusoids are congested.
Cardiac cirrhosis.
11. Rt Heart Failure
Congestive splenomegaly, marked sinusoidal dilatation.
Chronic intestinal oedema may result in malabsorption
syndrome.
Kidneys show congestion, fluid retention.
Peripheral oedema and azotemia as terminal event.
Brain shows hypoxia and increased congestion.
Pleural and pericardial fluid accumulation.
Dependent oedema and anasarca.
12. Left Sided Heart Failure
Causes are IHD, hypertension, aortic and mitral valve
disease, myocardial diseases.
There is progressive damming of blood in the
pulmonary circulation and diminished peripheral
pressure and flow.
Findings depend on cause; Left ventricle may
hypertrophied and/or dilated, fibrosis of myocardium,
left atrial enlargement and fibrillation, thrombosis,
congested lungs which appear heavy and wet,
kidneys and brain are congested due to retrograde
pressure.
13. Microscopic findings: perivascular and interstitial transudates,
Kerley B lines in lungs due to interlobular fluid. Progressive
oedematous widening of alveolar septa, accumulation of fluid in
the alveoli and later hemosiderin laden macrophages called
heart failure cells, dyspnoea, orthopnoea, paroxysmal nocturnal
dyspnoea, cough with expectoration.
Kidney activate renin angiotensin aldosterone mechanism due
to decreased perfusion, retention of sodium and water, blood
volume increases which aggravates pulmonary oedema, pre
renal azotemia.
Brain shows hypoxic encephalopathy.
15. Ischemic Heart Disease
Results from imbalance between perfusion and demand; Result
in decreased oxygen and nutrition supply and waste removal.
Isolated hypoxemia e.g. cyanosis, congenital disease, anemia
are not so damaging to heart as IHD.
Aetiology: coronary obstruction most likely due to
atherosclerosis, symptomatic after long periods.
Aggravating factors: hypertrophy, low BP, hypoxemia, increased
heart rate.
Onset of symptoms not predictable on the basis of severity of
atherosclerotic narrowing.
Young man with modest narrowing may land with acute
myocardial infarction.
Erosions, ulcerations, fissuring, rupture, deep hemorrhage and
superimposed thrombosis.
16. Syndromes of Myocardial Ischemia
Myocardial infarction: death of myocardium
Angina pectoris: stable, Prinzmetal, unstable
Chronic ischemia leading to heart failure
Sudden cardiac death
17. Epidemiology
Leading cause of death in men and women all over
the world.
Recent decrease in death incidence may be due to
preventive measures, therapeutic measures, various
medications, angioplasty, coronary by pass
operations, control of diabetes, administration of post
menopausal estrogens in females, lipid lowering
agents, aspirin and antioxidants.
18. Pathogenesis
Vast majority result from atherosclerotic narrowing
along with other interactions including vasospasms,
thrombosis and platelet aggregates.
More than 90% symptomatic cases show at least.
75 % reduction in the coronary blood flow.
Usually first a few cm (epicardial parts) of two or all
the three vessels are significantly narrowed.
In addition to fixed obstruction, plaque change may
precipitate the onset of sudden ischemia.
Vasoconstriction at the site of coronary obstruction
may be stimulated by adrenergic agonists, decreased
nitric oxide by endothelial cells and mediators from
mast cells.
19. Angina Pectoris
It is a symptom complex of IHD characterized by
paroxysmal and usually by recurrent attacks of
retrosternal chest pain precipitated by physical
exertion, emotional excitement, relieved by rest or
vasodilators like nitroglycerin.
20. Myocardial Infarction
It is the most important form of IHD.
Transmural vs. Subendocardial infarction.
Most are transmural resulting from chronic coronary
atherosclerosis with superimposed thrombosis.
Subendocardium is the most vulnerable part.
Transmural also starts as subendocardial.
21. Incidence and Risk Factors
Increasing age, hypertension, smoking, diabetes,
hypercholesterolemia. Males at any age are at
increased risk.
No race differences.
In post menopausal women estrogen therapy may
decrease the incidence.
22. Pathogenesis
Coronary artery occlusion.
Atherosclerosis.
Disruption manifested by hemorrhage, erosion or
ulcerations.
Subendothelial exposure of collagen and necrotic plaque
contents, platelet adhesion, aggregation, activation and
release of potent aggregators.
Activation of coagulation pathways.
Thrombus suddenly occludes the lumen of coronary artery.
In 10%, vasospasm, emboli, vasculitis, hemoglobinopathies
and other diseases of intramural portions of coronaries.
23. Myocardial Response
Coronary blood flow occlusion results in profound functional,
biochemical and morphological consequences.
Occlusion of a major coronary artery results in ischemia and cell
death in the anatomic region supplied by the vessel esp. At risk
is the subendothelial myocardium.
Outcome depends on the severity and duration of flow
deprivation. Within 60 sec, profound loss of contractility occurs.
Biochemical changes; Cessation of aerobic glycolysis,
decreased ATP and accumulation of lactic acid.
Early ultrastructural changes following ischemia are reversible in
20 to 40 minutes.
Myocardial ischemia also contributes to arrhythmias.
Sudden death in cardiac ischemia may be due to ventricular
fibrillation.
24. Time of Onset of Events in Ischemia
Onset of ATP depletion occurs in seconds.
Loss of contractility within 2 min.
Marked reduction of ATP occurs in 10 to 40 min.
Irreversible cell injury occurs in 20-40 min.
Microvascular injury follows in more than 60 min.
25. Morphological Changes.
Coagulative necrosis follows prolonged myocardial
ischemia 6-12 hours.
All transmural infarcts involve Lt ventricle and part of
septum; In only 3 % cases Rt ventricle alone is
involved.
Transmural infarcts encompass the entire perfusion
zone of the occluded artery with preservation of 0.1
cm of endocardial myocardium.
26. Macroscopic or Naked Eye Changes.
12-24 hours: dark mottling with nuclear pyknosis.
1-3 days: mottling with yellow tan infarct in the centre.
3-7 days: hyperemic border, central yellow tan softening.
7-10 days: yellow tan infarct, red tan margins.
2-8 weeks: Grey white scar.
Areas of ischemic damage undergo a progressive sequence of
changes consisting of coagulative necrosis followed by
inflammation and repair.
At autopsy the gross changes may be unapparent in 2-3 hours,
so tissue slices are immersed in triphenyltetrazolium chloride
which imparts, brick red colour to the surrounding viable
myocardium with unstained infarct area.
27. Microscopic Changes.
Routine tissue stains show detectable coagulative
necrosis in 4-12 hours. Surrounding myocardial cells
may show reversible cell injury especially in the
subendocardial regions.
The necrotic muscle elicits acute inflammation in 2-3
days. Macrophages remove the necrotic debris. Then
damaged area is progressively replaced by ingrowth
of highly vascularized granulation tissue.
Fibrous tissue replaces the granulation tissue in 2-4
weeks time.
Larger lesion may take longer times for final healing.
46. Infarct Modification After Reperfusion
Thrombolytic therapy, angioplasty or coronary
bypass.
Streptokinase or tissue type plasminogen activators
are used as thrombolytic agents, which activate the
human thrombolytic system. The artery may get
recanalized partly thereby limiting the infarct size,
with consequent improvement in both short and long
term function and survival. To achieve the purpose,
the time is very crucial.
Reperfusion within 15-20 min can prevent necrosis
but if longer times elapse, necrosis cannot be
prevented but viable myocytes can be salvaged.
47. . A complete infarct after reperfusion may be hemorrhagic due to
leaky vessels. Infarcted myocardium after reperfusion,
microscopically shows necrosis with contraction bands which
are intensely eosinophilic transverse bands composed of
closely packed hypercontracted sarcomeres. After reperfusion,
there is exaggerated contraction of myofibrils. Reperfusion
injury may also be initiated by oxygen free radicals.
Although most of the viable myocardium at the time of reperfusion
ultimately recovers, some sort of biochemical and functional
defect may remain for several days so called prolonged post
ischemic ventricular dysfunction.
Chronic silent myocardial ischemia may prevent the heart from a
greater ischemic attack by pre conditioning.
48. Signs and Symptoms
Sudden severe retrosternal pain, sweating, weak rapid pulse,
breathlessness, may be asymptomatic in 15% cases, esp.
among diabetics and very old persons..
Laboratory findings are based on measurements of various
macromolecules released in to the circulation from injured
myocytes.
Creatine kinase (CK) is an enzymes present in myocardium,
skeletal muscle and brain. Its isoenzymes are CK MM, CK BB
and CK MB. CK MB is concentrated in the myocardium. Total
CK levels begins to rise within first 2-4 hours, peak at 18 and
begin to fall after 72 hours time after onset of acute MI.
Measurement of CK MB activity is more specific because, CK
may also come from skeletal muscle injury.
49. Clinical Features Cont.
Other laboratory investigations include measurement of AST
and LDH activities.
LDH is released more slowly; Therefore it may not be as useful
as early markers of acute attack.
Another cardiac specific marker is estimation of protein
troponins (troponin 1 and troponin T) which are not detectable
normally.
After acute MI, levels are detectable at the same time as CK
MB. The levels remain elevated for 7-10 days.
Other diagnostic tools include electrocardiography,
radionucleotide angiography, perfusion scintigraphy and MRI.
50. Complications of MI
Death rate from acute MI has decreased from 30 to
15 % on accounts of impressive management and
reperfusion therapy. Half of these deaths occur within
1 hour, and these individuals never reach the
hospital.
Factors associated with poor prognosis include; old
age,female gender, diabetes and history of previous
attacks of MI.
Complications include;
Cardiogenic shock: have 70% mortality.
Arrhythmias: bradycardia, tachycardia, ventricular fibrillation
and asystole.
51. Complications Cont.
Myocardial Rupture; of wall and papillary muscle and
rupture of septum.
Pericarditis: fibrinous or fibrinohemorrhagic develops
after 2-3 days of transmural infarct which is self
limited.
Right ventricular infarction leads to serious functional
complications; although pure right sided disease is
rare.
Infarct extension.
Infarct expansion.
Mural thrombus: due to endocardial injury and loss of
myocardial contractility and stasis.
52. Complications
Ventricular aneurysms: late complication.
Papillary muscle dysfunction: mitral regurgitation.
Progressive heart failure:
Prognosis and development of complications
depends on: infarct size and transmural extent.
Large transmural infarcts develop cardiogenic shock,
arrhythmias and congestive cardiac failure.
Anterior transmural infarcts develop rupture, expansion,
mural thrombi and aneurysms; thus have the poor
prognosis.
Posterior transmural infarcts can be complicated by
conduction defects and right ventricular failure.
53. Long Term Prognosis
Depends on residual left ventricular function and
extent of coronary obstruction in the viable
myocardium.
The overall total mortality is around 30% within one
year; 3-4% mortality among the survivors with each
passing year.
Attempts to prevent the mortality among such
persons is called secondary prevention.
54. Chronic Ischemic Heart Disease
Mostly elderly people that develop progressive heart
failure following ischemic damage.
Usually constitutes post infarction cardiac
decompensation; In other cases severe obstructive
coronary disease may be present without acute or
healed infarction but with diffuse myocardial
dysfunction.
Congestive cardiomyopathy may be used by
clinicians who develop congestive cardiac failure with
past episodes of MI or anginal attacks.
55. Sudden Cardiac Death
Defined as unexpected death from cardiac causes
early within 1 hour after or without the onset of
symptoms.
Non atherosclerotic causes include: congenital
structural abnormalities, aortic valve stenosis,
prolapse of mitral valve, myocarditis, hypertrophic
cardiomyopathy, pulmonary hypertension,
abnormalities in conduction system.
56. Hypertensive Heart Disease, Left Sided
Response of the heart to increased pressure in the
systemic circulation.
Concentric hypertrophy develops as a compensatory
phenomenon which may lead to cardiac dilatation,
CCF and other cardiac dysfunctions.
Morphology: initially there may be circumferential
hypertrophy without increase in heart size, may
exceed above 2 cm, weight may increase > 500 gm.
Microscopically: increased transverse myocyte
diameter, followed by increased size variation and
interstitial fibrosis.
57. Clinical Aspects
Compensated hypertension may be asymptomatic,
detected only on ECG or echocardiography.
At times patient may present with atrial fibrillation or
congestive cardiac failure.
Other complications include atherosclerosis with
associated MI and CVA.
58. Pulmonary Hypertension
Also called cor pulmonale, constitutes right
ventricular hypertrophy, dilatation and failure
secondary to lung diseases or of pulmonary
vasculature.
Acute cor pulmonale develops following massive
pulmonary embolism.
Chronic is secondary to obstruction of pulmonary
arteries or arterioles or compression of septal
capillaries like in emphysema.
59. Morphology
Acute: marked right ventricular dilatation without
hypertrophy.
Chronic: ventricular wall thickness may be up to 1
cm.
Sometimes there may be tricuspid regurgitation with
fibrous thickening of the valve.
60. Valvular Heart Disease
May cause stenosis or incompetence.
These abnormalities may be pure or mixed.
Defect may involve single valve (isolated disease) or multiple
(combined).
Functional regurgitation results when ventricle dilates due to any
disease.
Valvular dysfunction may be slight or severe, that may be
rapidly fatal e.G. Infective endocarditis may destroy aortic valve,
resulting in rapidly progressing cardiac failure; In contrast,
rheumatic heart disease may be well tolerated due to insidious
onset.
Depending on degree, duration and cause, secondary changes
in heart, blood vessels and other organs may develop.
61. Causes of Acquired Valvular Dysfunctions
Mitral stenosis: rheumatic heart disease.
Mitral regurgitation.
Abnormalities of leaflets and commissures e.g.
postinflammatory scarring, infective endocarditis, mitral
valve prolapse.
Abnormalities of tensor apparatus e.g. Rupture of papillary
muscle or of chordae tendineae.
Abnormalities of left ventricular cavity and/or annulus e.g.
Left ventricle enlargement and calcification of mitral valve.
Aortic stenosis.
Post inflammatory scarring e.g. Rheumatic heart disease.
Senile calcific aortic stenosis.
62. Causes Cont.
Aortic regurgitation
Intrinsic valvular disease e.g. Postinflammatory scarring and
infective endocarditis
Aortic disease e.g. Degenerative aortic dilatation, syphilitic
aortitis, ankylosing spondylitis, rheumatoid arthritis and
Marfans syndrome.
63. Calcific Aortic Stenosis
Patients present in their 70’s or 80’s, as they had
congenitally normal valves.
Morphologic hall mark of non rheumatic calcific aortic
stenosis is heaped up calcified masses within aortic
cusps, preventing the opening of the valve.
Calcification involves the bases of the cusps.
Early process that is not important hemodynamically,
is called aortic valve sclerosis.
Mitral valve is not affected simultaneously, in contrast
to rheumatic heart disease.
64. Clinical Features of Aortic Stenosis.
It is the most common of all the valvular
abnormalities.
Due to obstruction to out flow pressure gradient may
exceed well above between left ventricle and aorta,
which results in hypertrophy of left ventricle.
Patient may present with angina or syncope,
mechanism is not known.
Onset of symptoms heralds the failure of
compensatory mechanism.
Death may result from left sided heart failure within 3
years if untreated.
65. Myxomatous Degeneration of
Mitral Valve
Affects 3% of adults usually young women.
One or both leaflets are enlarged, hooded, redundant
or floppy and prolapse back in to left atrium. Serious
complications develop in a minority of cases.
Morphology: affected leaflets are thick and rubbery,
tendinous cords are elongated, thinned and may be
ruptured, annular dilatation is characteristic.
Concomitant tricuspid involvement is 20-40%.
Histologically, attenuation of fibrosa layer of the
valve, on which the structural integrity of valve
depends along with focally marked thickening of the
spongiosa layer with deposition of mucoid material.
66. Morphology Cont.
Secondary changes include fibrous thickening of
valves esp. of rubbing surfaces, linear fibrous
thickening of left ventricular endocardium, thickening
of left atrial endocardium as a result of prolapsing
valves, thrombi on atrial surfaces of leaflets and focal
calcifications.
67. Pathogenesis and Clinical Features.
Developmental abnormality of connective tissue because in
Marfans syndrome, involvement of other systems is also seen,
caused by mutations in gene encoding fibrillin. Subtle defects in
structural proteins may predispose to damage by long standing
hemodynamic stress.
Most patients remain asymptomatic, there may be mid diastolic
click due to tensing of everted cusp, there may be holosystolic
murmur if regurgitation occurs.
Echocardiography reveals valve prolapse.
Chest pain, dyspnoea, fatigue, psychiatric disorders like
depression and personality disorders.
Serious complications include: infective endocarditis, mitral
insufficiency, thrombo embolism and arrhythmias.
69. Rheumatic Heart Disease
Rheumatic fever: acute immunologically mediated
multisystem inflammatory disease, a few weeks after
acute episode of beta hemolytic streptococcal
infection (throat). Acute rheumatic carditis may
progress to chronic valvular deformities. Recently
there is significant decrease in the complications of
this disease on account of raised socioeconomic
standards, better diagnostic facilities and early
treatment of pharyngitis.
Only 3% of group A beta streptococci develop this
serious complication.
70. Morphological Features
Acute rheumatic fever: disseminated but focal lesions
esp. in heart called Aschoff bodies, fibrinoid
degeneration surrounded by lymphocytes, plasma
cells and plump macrophages called Anitschkow cells
or caterpillar cells, some times multinucleated giant
cells may also form.
During acute attack, all the three layers of heart may
be involved- called pancarditis.
In pericardium, serofibrinous or fibrinous pericarditis.
Myocardium: scattered Aschoff bodies, called
myocarditis within interstitial connective tissue.
71. Morphology Cont.
Involvement of endocardium and left sided valves by
inflammatory foci comprises of fibrinoid necrosis
within cusps or along the tendinous cords on which
small vegetations called verrucae along the lines of
closure of valves.
Warty projections are due to precipitation of fibrin at
the sites of erosions related to underlying
inflammation and degenerations.
These changes in acute fever cause no disturbances
to valvular functions.
72. Morphology Cont.
Subendocardial lesions in left atrium are called
MacCallum plaques.
Chronic rheumatic heart disease: organization of
acute inflammation and deforming fibrosis e.G.
Thickening and retracted valve with fusion of leaflets
causing permanent deformity and narrowing,
shortening, thickening and fusion of tendinous cords;
Mitral and aortic valves are most important.
Microscopically: diffuse fibrosis and
neovascularization replacing the original avascular
leaflets, Aschoff bodies not present in ch. Disease.
73. Morphology Cont.
Rheumatic heart disease is the most frequent cause
of mitral stenosis.
Mitral valve alone is involved in 65-70% cases of
rheumatic disease.
Tricuspid valve is involved less frequently and with
much less severity.
If there is calcification or formation of fibrous bridges
across the valvular commissures, buttonhole or fish
mouth stenosis.
Lt atrial hypertrophy, dilatation, pulmonary
congestion, pulmonary hypertension and right
ventricular hypertrophy.
Normal left ventricle in pure mitral valve stenosis.
74. Pathogenesis and Clinical Features.
Hypersensitivity reaction induced by group A
streptococci.
Antibodies directed against M proteins of certain
strains cross react with tissue glycoproteins in heart,
joints and other tissues.
Genetic susceptibility regulates the hypersensitivity
reaction because minority of patients develop this
complication.
Children 5-15 yrs.
By the time fever develops, throat cultures are
negative but antibodies to streptolysin o and anti
DNAse are usually detectable in sera.
75. Clinical Features Cont.
Arthritis and carditis predominant clinical features.
Arthritis is migratory polyarthritis, with fever, mostly
large joints are involved.
Features of carditis include: pericardial friction rub,
weak heart sounds, tachycardia and arrhythmias.
Usually no mortality in acute disease.
After initial attack, there is increased vulnerability to
reactivation of disease with subsequent pharyngitis.
Carditis worsens with each attack and damage is
cumulative.
Other hazards are embolism and infective
endocarditis.
76. Clinical Features Cont.
Chronic rheumatic carditis is not manifested clinically
for years of decades after initial acute attack.
Signs and symptoms depend upon the valves
involved.
Cardiac murmurs, cardiac hypertrophy and dilatation,
heart failure, arrhythmias like atrial fibrillation,
thrombo embolic phenomenon and infective
endocarditis.
Replacement of mitral valve with prosthetic devices
has greatly improved the prognosis.
77. Infective Endocarditis
Serious disease characterized by colonization of
cardiac valves, endocardium and other
cardiovascular sites by a microorganism.
Bulky friable vegetations composed of thrombotic
debris along with organisms with destruction of
underlying cardiac tissues.
Aorta, aneurysmal sacs, other blood vessels and
prosthetic valves may be infected.
Microorganisms are bacterial, fungi, rickettsiae and
chlamydia. Most important are bacteria-bacterial
endocarditis.
78. Classification
Clinically divided in to acute and subacute depending
on virulence of the causative organisms and
presence of underlying cardiac disease.
Acute: destructive infection of a previously normal
heart valves by a highly virulent organism, that can
lead to death of around 50% of the patients within
days to weeks in spite of intensive management.
Subacute: caused by organisms of low virulence
affecting previously abnormal heart, particularly
deformed valves. The disease runs protracted course
and may settle with appropriate treatment.
79. However both clinical and morphologic patterns
suggest, possibility of a spectrum rather than a clear
cut demarcation between acute and subacute.
80. Pathogenesis
Usually cardiac and vascular abnormalities
predispose to this form of infection.
Previously rheumatic heart disease was major
contributor but recently, myxomatous mitral,
degenerative calcific valvular stenosis, biscuspid
aortic valve, prosthetic valves and vascular grafts are
more common.
Predisposing host factors include: neutropenia,
immunodeficiency, therapeutic immunosuppressive
drugs, diabetes and alcohol and intravenous drug
abuse. Indwelling cardiac or vascular catheters may
also cause infective endocarditis.
81. In 50-60% of the cases, the causative organisms are
alpha hemolytic streptococci called viridans
streptococci.
10-20% cases are caused by staphylococcus aureus,
being major offenders in intravenous drug abusers.
Other bacteria include: enterococci, Haemophilus,
Acinetobacteria, Cardiobacterium; Commensals of
oral cavity.
Prosthetic valve carditis is most often caused by
coagulase negative staphylococci.
Other agents: gram negative bacilli and fungi.
82. In 10% cases no offending agent can be isolated
called culture negative endocarditis, may due to
previous antimicrobial treatment.
Portal of entry of microorganisms may be an obvious
focus of infection such as a dental or other surgical
procedure that causes a bacteremia, intravenous
injection of bacteria by I/V drug users, or from an
occult source from GIT, oral cavity or trivial injuries.
83. Morphologic Features.
Both acute and subacute develop friable, bulky
vegetations, containing fibrin, inflammatory cells and
bacteria, most commonly on heart valves, most often
mitral and aortic. Right sided valves may be involved
in I/V drug abusers.
Vegetations may be single or more often multiple.
Vegetations are often destructive to underlying
myocardium producing abscess cavity.
Fungal vegetations are usually larger.
In subacute form, vegetations are smaller and less
destructive.
84. Morphology Cont.
Systemic embolism may occur at any time because
of friability of the vegetations; Infarcts in brain,
kidneys, myocardium etc, septic infarcts may cause
abscesses in the above said tissues.
Microscopically vegetations consist of granulation
tissue at their bases; With the passage of time,
fibrosis, calcifications and chronic inflammatory
infiltrate may develop.
85. Clinical Features
Fever most important feature, may be absent in
elderly.
Non specific fatigue, loss of weight and flu like state.
Murmurs in 90% cases.
Features like petechial rashes, subungual
hemorrhages and Roth spots in eyes have become
less common because of effective antimicrobials.
Acute disease has stormy course, high grade fever
with chills, weakness and lassitude.
Cardiac complications: stenosis or insufficiency,
myocardial ring abscesses and suppurative
pericarditis.
86. Embolic complications: cerebral infarcts, meningitis,
myocardial infarction,kidney abscesses, lung infarcts
and abscesses, pneumonia.
Focal and diffuse glomerulonephritis leading to
hematuria, albuminuria and renal failure.
Splinter hemorrhages in the nail beds.
92. Non Infected Vegetations
Non bacterial thrombotic endocarditis are
characterized by deposition of small masses of fibrin,
platelets and other blood components on the valvular
leaflets in debilitated patients. The vegetations are
sterile and loosely attached resulting in embolism.
93. Myocardial Diseases.
Inflammatory disorders, immunologic diseases,
systemic metabolic disorders, muscular dystrophies,
genetic abnormalities of myocardial cells and
diseases of unknown cause.
Cardiomyopathies are a group of diseases resulting
from a primary abnormality in the myocardium.
Clinicopathologic patterns are: dilated
cardiomyopathy, hypertrophic and restrictive
cardiomyopathy.
Of these dilated one is most common (90%).
Each of these may be idiopathic or specific cause.