2. INTRODUCTION
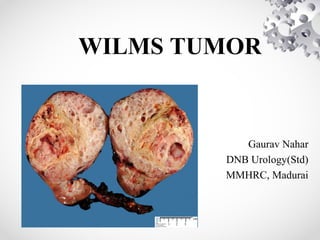
• Wilms tumor - Nephroblastoma.
• Most common primary malignant renal tumor
of childhood.
• This embryonal tumor develops from remnants
of immature kidney.
3. EPIDEMIOLOGY
• Accounts for 6% to 7% of all childhood
cancers.
• Children<15 yrs: annual incidence rate 7 to 10
cases per million.
• More than 80% of cases are diagnosed before
5 years of age, with a median age of 3.5 years.
4. • B/L Wilms tumors present at earlier age.
• Incidence lower in East Asian populations &
higher in black populations compared with
North American and European whites.
• Environmental risk factors play a minor role.
5. ETIOLOGY
• Majority of Wilms tumors arise from somatic
mutations restricted to tumor tissue.
• A much smaller percentage originate from
germline mutations.
• Several genetic events result in Wilms tumor
development.
6. • 10% tumors- have coexistent congenital
anomalies and syndromes.
• 5% to 10% tumors- bilateral/multicentric.
• 1% to 2% are familial.
7.
8. WT1:
• 1st Wilms tumor gene to be identified.
• Gross deletions at chromosome 11p13.
• Associated syndromes:
1.WAGR syndrome
2.Denys-Drash syndrome
3.Frasier syndrome
9. • WT1 gene is important for normal kidney &
gonadal development.
• WT1 encodes a zinc-finger transcription factor
expressed in kidney, gonads, spleen, &
mesothelium
• WT1 is necessary for ureteric bud outgrowth
and nephrogenesis.
10. • WAGR (Wilms tumor, Aniridia, Genital
anomalies, mental Retardation) syndrome.
• Aniridia, found in 1.1% of Wilms tumor
patients, is caused by an abnormality of the
PAX6 gene located adjacent to WT1.
• Wilms tumor develops in 40% to 70% of
aniridia patients with deletions of WT1.
11. • Denys-Drash syndrome (DDS): specific
association of male pseudohermaphroditism,
renal mesangial sclerosis, and nephroblastoma.
• Caused by point mutations in zinc finger DNA
binding region of WT1.
• >90% of DDS patients harbor germline point
mutations in only one WT1 allele.
12. • WAGR and DDS patients- more likely to have
bilateral tumors & are diagnosed at a younger age.
• WAGR patients- increased risk of renal failure if they
survive into puberty.
• Genitourinary anomalies- renal fusion anomalies,
cryptorchidism, hypospadias are present in 4.5% of
patients with Wilms tumor.
14. • Beckwith-Wiedemann syndrome(BWS)
characterized by excess growth at cellular,
organ (macroglossia, nephromegaly,
hepatomegaly), or body segment
(hemihypertrophy) levels.
• Adrenocortical neoplasms and hepatoblastoma
also occur in BWS.
• Most cases sporadic; 15% heritable- AD.
15. WTX:
• Tumor suppressor gene, Wilms Tumor gene on
the X chromosome, at Xq11.1,
• Inactivated in up to one third of Wilm's
tumors.
• Targets single X chromosome in males &
active X chromosome in females with tumors.
16. Familial Wilm's Tumor : (FWT1, FWT2)
• 1% to 2% of Wilms tumor patients have a
family h/o Wilms tumor.
• Earlier age of onset & increased frequency of
B/L disease.
17. • TP53 mutations- increased frequency in
anaplastic histology.
• LoH at 1p and 16q are associated with an
increased risk of tumor relapse and death.
18. SCREENING
• Ultrasound surveillance- from time of
diagnosis until 5 years of age, with a
frequency of every 3 to 4 months.
• BWS, Simpson-Golabi-Behmel, and familial
Wilms- continue to 7 years.
• Screening recommended when WT incidence
> 5%.
• Screening of contralateral kidney after
nephrectomy for U/L Wilm's.
19. • CT or MRI if USG shows any suspicious lesion.
• 7-fold increased risk of Wilm's tumor in HK.
• an increased risk of müllerian duct anomalies in girls
with Wilms tumor- Approx.10% girls will have
abnormalities, such as duplication of cervix or uterus,
or bicornuate uterus.
20. PATHOLOGY
Favorable-Histology Wilms Tumor(FH):
• Wilms tumor compresses adjacent normal
renal parenchyma, forming an "intrarenal
pseudocapsule."
• Tremendous histologic diversity.
• 90% of all renal tumors have favorable
histology.
21. • “Classical” Wilms tumor is characterized by
islands of compact undifferentiated blastema,
presence of variable epithelial differentiation
in the form of embryonic tubules, rosettes, and
glomeruloid structures,
separated by a significant stromal component.
22.
23. • Predominantly epithelial differentiation- low
degree of aggressiveness, majority are stage I
tumors,
• But may be more resistant to therapy, if they
present as advanced-stage disease.
25. Anaplastic Wilms Tumor:
• Anaplasia is characterized by the presence of
three abnormalities:
1.nuclear enlargement to three or more times the
diameter of adjacent cells,
2.hyperchromasia of enlarged nuclei, and
3.abnormal mitotic figures.
• Rarely seen in children< 3 years.
26. • Resistant to chemotherapy.
• Poor prognosis.
• Further divided into focal & diffuse patterns.
27. Nephrogenis Rests:
• Precursor lesions; still most don't form Wilm's
tumor.
• A rest can undergo maturation, sclerosis,
involution, or complete disappearance.
• Two types based on location: Perilobar &
Intralobar(PLNRs & ILNRs).
28. • Perilobar NRs- found only in the lobar
periphery, elaborated late in embryogenesis.
• Subcortical, sharply demarcated, and contain
predominantly blastema & tubules.
• Usually found in BWS, linked to 11p15 locus.
29. • Intralobar NRs found anywhere within the
lobe, renal sinus and wall of PCS.
• Result of earlier gestational aberrations.
• ILNRs are commonly stroma rich.
• Typically seen in aniridia, WAGR, DDS or
other features a/w WT1.
30.
31. CLINICAL PRESENTATION
• A palpable smooth abdominal mass- 90%.
Incidentally discovered.
• Abdominal pain, gross hematuria & fever- less
frequent.
• Tumor rupture with hemorrhage into peritoneal
cavity- mimics acute abdomen.
• Extension into renal vein & IVC- varicocoele,
hepatomegaly due to hepatic vein obstruction, ascites,
and congestive heart failure- <10%.
32. • Hypertension- common at diagnosis, d/t
elevated plasma renin levels; resolves shortly
after removal.
• Acquired von Willebrand disease found in 8%
of newly diagnosed Wilms tumor.
33. IMAGING
• FOUR FIELD CHEST RADIOGRAPHY:
may show lung metastasis.
RENAL ULTRASOUND:
1st study to evaluate child with abd.mass.
demonstrate solid nature of the lesion.
Doppler USG helps to exclude intracaval
tumor extension, & its proximal extent.
34. Solid renal tumor: CT demonstrates that lesion is amenable to renal-sparing
surgery
35. • CT SCAN:
helps to determine origin of the tumor, lymph
node involvement, B/L kidney involvement,
invasion into major vessels (IVC), and liver
metastases.
CT chest to rule out lung metastasis.
36. CT scan of a left Wilms tumor with a small rim of
functioning renal parenchyma
37. • MRI ABDOMEN:
Most sensitive imaging modality for caval
patency, to determine tumor extension into
IVC.
low signal intensity on T1-weighted images
and high signal intensity on T2-weighted
images.
40. DIFFERENTIAL DIAGNOSIS
• Mesoblastic nephroma - Most common renal tumor in the
first month of life.
• Renal cell carcinoma
• Clear cell sarcoma of the kidney
• Rhabdoid tumor of the kidney
• Nonmalignant mass
• Hydronephrosis
• Multicystic kidney disease
• Renal cyst
• Renal thrombosis
• Dysplastic kidney
• Renal hemorrhage
42. PROGNOSTIC FACTORS
• Most Important determinants of outcome:
histopathology & tumor stage.
• Chromosomal Abnormalities: LOH for
chromosome 16q and/or 1p (20% of Wilms
tumors) a/w increased risk for relapse & death.
• High telomerase activity- an unfavourable
prognostic feature.
• DNA Content: Aneuploidy & DNA index .
1.5- strongly a/w anaplastic histology.
• Cytokines: VEGF angiogenic cytokine.
43. TREAMENT
• Usual approach- nephrectomy followed by
chemotherapy, with or without postoperative
radiotherapy.
• Multiple RCTs to determine therapeutic protocols by:
1. National Wilm's Tumor Study Group/Children's
Oncology Group(NWTSG/COG),
2. International Society of Pediatric Oncology(SIOP),
and
3. United Kingdom Children’s Cancer Study Group
(UKCCSG) .
44.
45. COG AREN0321 protocol for high risk Wilms
tumor
• Focal anaplastic stage I-III Wilms tumors and diffuse
anaplastic stage I Wilms tumors - Nephrectomy followed by
vincristine, actinomycin-D, and doxorubicin in addition to
local radiotherapy
• Focal anaplastic stage IV Wilms tumors and diffuse
anaplastic stage II-III tumors –Patients undergo the same
treatment, with the addition of cyclophosphamide, etoposide,
and carboplatin
• Stage IV diffuse anaplastic Wilms tumors - More
aggressive treatment is delivered; nephrectomy is followed by
initial irinotecan and vincristine administration, which in turn
is followed by actinomycin-D, doxorubicin,
cyclophosphamide, carboplatin, etoposide, and radiotherapy.
46. SURGICAL CONSIDERATIONS:
• Radical nephrectomy by transperitoneal approach.
• Thorough exploration of the abdominal cavity to
exclude local tumor extension, liver and nodal
metastases, or peritoneal seeding.
• Accurate staging to determine appropriate
chemotherapy & need for radiation therapy.
• Selective sampling of suspicious nodes is essential.
• Formal RPLND is not recommended.
47. • Risk factors for local tumor recurrence
(Shamberger):
1.tumor spillage,
2.unfavorable histology,
3.incomplete tumor removal, and
4.absence of any lymph node sampling.
48. PREOPERATIVE CHEMOTHERAPY:
• Situations where preoperative chemotherapy is
recommended:-
1. Children for whom renal-sparing surgery is planned,
2. Tumors are inoperable at surgical exploration, and
3. There is tumor extension into IVC above hepatic
veins.
• An inoperable tumor should be considered stage III
and treated accordingly.
49. • Inoperability should not be based on
preoperative imaging studies, which can
overestimate local tumor extension.
• Pretreatment with chemotherapy almost
always reduces the bulk of tumor and renders
it resectable.
• Majority of reduction in tumor volume occurs
in first 4 weeks of chemotherapy.
50. A, MRI of a Wilms tumor that was pretreated with chemotherapy.
B, After 6 weeks of chemotherapy, the tumor is much smaller in size
51. MANAGEMENT OF LUNG METASTASIS:
• CXR negative but CT positive: tissue
diagnosis of lung nodules because several
conditions (eg, histoplasmosis, atelectasis,
pseudotumor, intrapulmonary lymph node,
pneumonia) can mimic pulmonary metastases.
52. • WT FH with lung mets, no other mets/1p or
16q deletion: 6 weeks of actinomycin-D,
doxorubicin, and vincristine.
Complete resolution- No radiation required.
No resolution- cyclophosphamide and
etoposide in addition + radiation therapy.
53. MANAGEMENT OF B/L WILMS TUMORS:
• No initial radical nephrectomy.
• Preoperative chemotherapy for 6 weeks.
• tumors amenable to renal-sparing procedures
can proceed with surgery.
• Tumors not responding- B/L open biopsy &
additional chemo based on biopsy findings.
54. • Proceed to Sx at 12 weeks of therapy (no
benefit beyond 12 weeks).
• Partial nephrectomy, tumor enucleation or
wedge excision of tumor.
• In FH tumors, even with positive margins or
large B/L residual masses, adjuvant therapy
provides a good outcome.
55. LATE EFFECTS OF Rx
RADIATION:
• Musculoskeletal problems like scoliosis.
• Reduction in stature.
• Hypogonadism & temporary azoospermia.
• Delayed sexual maturation.
• Ovarian failure.
• Adverse pregnancy outcomes with increased
risk for miscarriage, LBW, prematurity &
congenital malformations.
• Increased risk of 2nd malignant neoplasms.