Contenu connexe
Similaire à Saudi arabia medical device regulatory process
Similaire à Saudi arabia medical device regulatory process (20)
Saudi arabia medical device regulatory process
- 1. Saudi Arabia
© 2016 Emergo – Have comments or suggestions about the content of this chart? Email us at marketing@emergogroup.com. Chart updated 05/2016. EmergoGroup.com/saudi-arabia
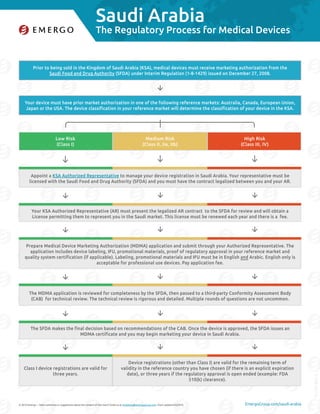
Prior to being sold in the Kingdom of Saudi Arabia (KSA), medical devices must receive marketing authorization from the
Saudi Food and Drug Authority (SFDA) under Interim Regulation (1-8-1429) issued on December 27, 2008.
The MDMA application is reviewed for completeness by the SFDA, then passed to a third-party Conformity Assessment Body
(CAB) for technical review. The technical review is rigorous and detailed. Multiple rounds of questions are not uncommon.
The SFDA makes the final decision based on recommendations of the CAB. Once the device is approved, the SFDA issues an
MDMA certificate and you may begin marketing your device in Saudi Arabia.
Your device must have prior market authorization in one of the following reference markets: Australia, Canada, European Union,
Japan or the USA. The device classification in your reference market will determine the classification of your device in the KSA.
Appoint a KSA Authorized Representative to manage your device registration in Saudi Arabia. Your representative must be
licensed with the Saudi Food and Drug Authority (SFDA) and you must have the contract legalized between you and your AR.
Your KSA Authorized Representative (AR) must present the legalized AR contract to the SFDA for review and will obtain a
License permitting them to represent you in the Saudi market. This license must be renewed each year and there is a fee.
Prepare Medical Device Marketing Authorization (MDMA) application and submit through your Authorized Representative. The
application includes device labeling, IFU, promotional materials, proof of regulatory approval in your reference market and
quality system certification (if applicable). Labeling, promotional materials and IFU must be in English and Arabic. English only is
acceptable for professional use devices. Pay application fee.
Low Risk
(Class I)
Class I device registrations are valid for
three years.
Medium Risk
(Class II, IIa, IIb)
Device registrations (other than Class I) are valid for the remaining term of
validity in the reference country you have chosen (if there is an explicit expiration
date), or three years if the regulatory approval is open ended (example: FDA
510(k) clearance).
High Risk
(Class III, IV)
5198-0516
The Regulatory Process for Medical Devices
- 2. Saudi Arabia
Notes
1. The time frames shown are typical for the majority of medical device submissions but assume that your device does not contain animal tissue or medicinal
substances. Your length of approval will depend on the quality and completeness of your technical documentation and how much time you take to address
additional information requests from the Conformity Assessment Body or SFDA after submission. YOUR SUBMISSION(S) MAY TAKE MORE TIME THAN
WHAT IS SHOWN ABOVE.
2. Registrations remain valid for the time specified as long as you do not make significant changes to the device design that result in reclassification,
intended use, or indications, or lose certification in the reference market. Higher risk devices will be valid for the remaining validity in the reference
country leveraged (if there is an explicit expiration date) or three years if the reference country approval is considered by the SFDA to be open ended.
3. Renewals should be initiated at least 2 months prior to expiration.
4. Our rating of the complexity of the registration process is based on our experience and the opinion of nearly 1,000 QA/RA professionals worldwide who
were asked to rate the difficulty of registering a device in each country. The European CE Marking process is considered the mid-point to which all other
markets are compared.
5. Low = Less than US$5,000; Midpoint = US$15,000-$30,000: High = More than US$50,000. Overall cost includes registration application fees, product
testing, in-country representation, submission preparation consulting and translation of registration documents but not IFU. Costs assume you already
have approval for your device in Australia, Europe, Canada, Japan or the United States.
EmergoGroup.com/saudi-arabia
Device classification
category
Low Risk
(Class I)
Moderate Risk
(Class II, IIa, IIb)
High Risk
(Class III, IV)
How long you should
expect to wait after
submission until
approval is granted1
1-6 months 1-6 months 1-6 months
Validity period for
device registrations2 3 years Up to 3 years Up to 3 years
Registration renewal
should be started this
far in advance3
2+ months 2+ months 2+ months
Complexity of the
registration process
for this classification4
Overall cost of gaining
regulatory approval5
5198-0516
Low High
Simple Complex
Low High
Simple Complex
Low High
Simple Complex
© 2016 Emergo – Have comments or suggestions about the content of this table? Email us at marketing@emergogroup.com. Table updated 05/2016.
Time, Cost, and Complexity of Registration