![BASES BIOQUÍMICAS DEL METABOLISMO ENERGÉTICO ,[object Object],[object Object],[object Object],[object Object],[object Object]](data:image/gif;base64,R0lGODlhAQABAIAAAAAAAP///yH5BAEAAAAALAAAAAABAAEAAAIBRAA7)
Recommandé
Contenu connexe
Tendances
Tendances (20)
En vedette
En vedette (20)
Similaire à Carbohidratos
Similaire à Carbohidratos (20)
Plus de rosalba alfonso
Plus de rosalba alfonso (19)
Dernier
Dernier (12)
Carbohidratos
- 1. BASES BIOQUÍMICAS DEL METABOLISMO ENERGÉTICO - CARBOHIDRATOS
- 3. BASES BIOQUÍMICAS DEL METABOLISMO ENERGÉTICO Y PROTEICO METABOLISMO Reacciones químicas - transforman materia y energía Catálisis enzimática – sucesión reacciones = vía metabólica Metabolitos e intermediarios Catabolismo : rompimiento moléculas complejas - nutrientes Anabolismo : Moléculas complejas de simples . E – ATP (catabolismo) o NADPH ( corregido ) CATABOLISMO DE MOLÉCULAS DE COMBUSTIBLE (Hans Krebs) Etapa 1. Fraccionamiento - almidón, PROT y TGC: monosacaridos, AA, glicerol y ácidos grasos. Poca energía libre Etapa 2. moléculas simples catabolizadas a pocas moléculas posibles de oxidación a CO2 y H2O en una vía metabólica común. Algo de energía libre ATP Etapa 3. Vía común: ATC, TTE electrones, fosforilación oxidativa Oxidan Acetil-coenzima A a CO2 y H2O y atrapan E como ATP Competencia por enzimas y cofactores
- 6. DESTINO FINAL DEL METABOLISMO DE LA GLUCOSA Fuentes: digestión de almidón, sucrosa, maltosa y galactosa. Absorbida y fosforilada en el hígado – glucógeno o metabolizada para E: glucolítica o Krebs o grasa Fuentes glucosa: Exógena, hidrólisis de glucógeno del hígado y músculos, Gluconeogénesis (glucosa de fuentes diferentes de CH) Glucólisis: Fraccionamiento enzimático de glucosa en 2 piruvatos y convertimiento a lactato. Vía oxidativa, no necesita O2. Aerobia (6 moles de ATP X mol/glucosa) o anaerobia (2 ATP / molécula de glucosa) MAYORES VIAS METABOLICAS ….
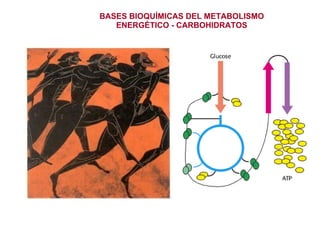
- 7. 1. GLUCOLISIS molécula de glucosa (6 carbonos) – oxidada = 2 moléculas de piruvato (3 carbonos) Energía almacenada como ATP (2 ATP/mol glucosa) Piruvato produce etanol, acetil-coenzima A, lactato. Músculo esquelético Citosol
- 8. carbohidratos, proteínas y grasas 2. Ciclo Krebs Mitocondria 1 Acetil CoA se oxida = 1 GTP + 4 electrones (3NADH, 1 FADH). Conduce síntesis de 9 moleculas ATP
- 9. 3. PENTOSA FOSFATO Citosol: descarboxilación oxidativa de glucosa 6-fosfato – 2 moléculas de NADPH para biosíntesis y formación de ribosa 5-fosfato para síntesis de nt. 4. SINTESIS Y DEGRADACIÓN DE GLICÓGENO Polimero de residuos de glucosa, una fosforilasa cataliza el clivaje por ortofosfato para producir 1-fosfato, glucosa 6-fosfato que entra al metabolismo
- 10. 5. Gluconeogénesis: glucosa desde aminoácidos. Riñón, hígado. Síntesis de glucosa desde lactato, glicerol y AA Punto entrada Piruvato - oxaloacetato
- 11. Cuerpos cetónicos: ahorro E músculo En el hígado alguna Acetil Coenzima A es convertida a cuerpos cetónicos ( acetoacetate, beta-hydroxybutyrate y acetone). Cetósis en inanición Acetil coenzima A derivada de B -oxidación de acidos grasos 6. SINTESIS Y DEGRADACIÓN DE ACIDOS GRASOS
- 12. COMPARTIMENTALIZACIÓN DE LAS MAYORES VIAS DEL METABOLISMO
- 13. RESUMEN: Interacciones claves de Glucose 6-phosphate, Pyruvate, and Acetyl CoA Entra a la célula y rápidamente es fosforilada a glucose 6-phosphate y almacenada como glicogeno, degradada a piruvato o convertida a ribose 5-phosphate. Se forma glycogeno cuando glucose 6-phosphate y el ATP son abundantes. Por el contrario, glucose 6-phosphate entra en la vía glucolítica cuando se requiere ATP o esqueletos de carbonof El otro camino de la glucose 6-phosphate, para fluir a través de la vía de la pentose phosphate, para proveer NADPH para biosíntesis reductiva y ribose 5-phosphate para la síntesis de nucleótidos. Glucose 6-phosphate se forma de la movilización del glucógeno o puede ser sintetizada de piruvato o AA glucogenicvía gluconeogénesis Glucose 6-phosphate .
- 14. Pyruvate se reduce a lactate por la lactate dehydrogenase para generar NAD +: glycolysis en condiciones anaerobicas del musculo. El lactate formado en tejidos activos es oxidado a piruvato nuevamente en otros tejidos. Transaminación de piruvato citosol a α -ketoacid, a alanine, el aminoácido correspondiente. Varios AA pueden ser convertidos a piruvato . La transaminación es el mayor link entre el metabolismo de CH y AA . Ocurre en citosol. Carboxylation a oxaloacetate en la mitocondria, primer paso de gluconeogenesis Decarboxylation oxidativa a acetyl CoA, irreversible, dentro mitocondria, reacción decisiva en el metabolismo; compromete los átomos de C de CH y AA a oxidación por el TCA o la síntesis de lipidos. Pyruvate : Es un α -ketoacid ide 3 carbonos. El Pyruvate se deriva primariamente de glucose 6-phosphate, alanine, y lactate.
- 15. La mayor fuente de sus dos carbonos activados son la decarboxilación del piruvato y la β-oxidation de los ácidos grasos. Acetyl CoA es también derivada de aminoácidos ketogénicos. La unidad acetil puede ser completamente oxidada a CO 2 por el ciclo del ácido cítrico. Acetyl CoA Alternativamente , 3-hydroxy-3-methylglutaryl CoA puede ser formada de 3 moléculas de acetyl CoA. Esta unidad de 6 carbonos es precursor de colesterol y cuerpos cetónicos, las cuales son formas de transporte de unidades de acetil liberadas del hígado para el uso por tejidos periféricos. La acetil CoA es exportada al citosol en forma de citrato para la síntesis de ácidos grasos.
- 17. DIABETES Conjunto de desórdenes que comparten la hiperglicemia: Reducida secreción insulina Disminución en el uso o incremento en la producción de glucosa Clasificación de acuerdo a proceso patológico que conlleva a hiperglicemia DM tipo II: grupo de desordenes con variable grado de resistencia a la insulina, alterada secreción e incremento en producción de glucosa
- 18. POR QUÉ SE SECRETA INSULINA POR CELULAS B PANCREAS
- 19. La glucosa y otros nutrientes regulan la secreción de insulina por las células B pancreáticas. Glucose es transportada por GLUT2 (glucose transporter); Seguido la glucosa es metabolizada por por las células B que alteran la actividad de canales iónicos llevando a la secreción de insulina. La secreción empieza con el transportador dentro de la célula B. El receptor SUR es un sitio de unión para drogas queactúan como secretores. SUR, sulfonylurea receptor; ATP, adenosine triphosphate; ADP, adenosine diphosphate. Glucose es el principal regulador de la secreción de insulina por células B pancreáticas, glucosa de >3.9 mmol/L (70 mg/dL) estimula síntesis de insulina: mejora procesamineto y sintesis. La fosforilación de glucosa por glucosinasa es el paso limitante que controla la secreción de insulina dependiente de glucosa. Metabolismo de glucosa 6-fosfato por glicolisis genera ATP que inhibe la actividad de un canal iónico de K+ ATP sensible. El canal consiste de dos proteínas separadas: uno es el receptor para ciertas hipoglucemias orales (e.g., sulfonylureas, meglitinides); el otro es un rectificador la proteína del canal de K+. Inhibición de el canal K+ induce despolarización de la membrana de células B que opera con voltage dependiente de canales de calcio (lllevando a un influjo de Ca+), y estimula la secreción de insulina. El patrón de secreción de insulina es pulsatil de liberación hormonal con un estallido secretorio cada 10 min, superimpuesto después de 80 a 150 min. Comida induce grandes incrementos 4 a 5 veces la línea base usualmente las últimas 2-3 h antes de retorno a línea base.
- 20. TRANSDUCCIÓN DE SEÑAL DE INSULINA EN EL MÚSCULO CITOESQUELÉTICO El receptor de insulina tiene actividad intrínseca tirosín kinasa e interactúa con el receptor de insulina sustrato (IRS) Diferentes proteínas se unen a estas proteínas celulares y se inicia la acción metabólica de la insulina. Insulina incrementa el transportador de glucosa vía PI-3 kinasa y la vía Cbl y promueve la traslocación intracelular de vesículas que contienen el transportador GLU4 a la membrana plasmática
- 21. Acción insulínica Una vez secretada al sistema porta, 50% es degradada por el hígado. Luego la insulina entra a circulación sistemica y se une a los receptores de los sitios blanco. Estimula actividad tirosin kinasa, el receptor se autofosforila y recluta moleculas y señales intracelulares, como insulina receptor sustrato (IRS). Estas y otras proteinas adaptadoras inician cacadas de fosfo y defosforilación ampliando el efecto mitogénico de la insulina, ejemplo, phosphatidylinositol-3'-kinase (PI-3-kinase) que estimula translocación de receptores (ej. GLUT4) a la superficie celular para la toma de glucosa en musculo y grasa. Por otra vía uinduce la síntesis de glucogeno, proteinas, lipogenesis y otros genes.
- 22. Señalización de la insulina
- 23. Incrementados niveles de lípidos en tejido muscular en diabetes tipo II, altera la oxidación glucosa e induce resistencia a la insulina (hígado) e interfiere con el metabolismo energético. El incrementado riesgo de diabetes asociado a obesidad, puede ser causado por acumulación muscular de lipidos creando resistencia a la insulina. Leptina, hormona secretada por el tejido adiposo asociada con muchos procesos, en hipotálamo regula alimento y metabolismo. La leptina en tejido músculo esquelético activa AMP-protein kinasa (AMP-kinasa), que tiene un papel importante evolutivo en señalización en repuesta a nutrientes. La AMPK fosforila e inactiva la ACC (acetyl-CoA carboxylase). La ACC catalisa la producción de malonyl-CoA desde acetyl-CoA. Malonyl-CoA es un inhibidor de ácidos grasos en la mitocondria por la carnitina palmitol transferasa I para oxidación y producción de energía. En presencia de leptina, la AMPK es activada, ACC es inhibida y caen los niveles de malonyl-CoA, incrementa la oxidación de ácidos grasos y reduce el contenido de lípidos de la célula. El contenido de lípidos reducido en el músculo citoesquelético permite la señalización insulina y el consumo de glucosa para retornar a niveles normales, reduciendo la resistencia a la insulina. Adiponectin facilita la sensibilidad a la insulina e inhibe pasos de procesos inflamatorios . En hígado inhibe la expresión de enzimas gluconeogénicas y la rata de producción de glucosa. En muscle, incrementa el transporte de glucosa, mejora oxidaciópn de ácidos grasos parcialmente debida a kinase. Resistencia a la insulina:
- 24. Resistencia a la insulina: Fosforila e inactiva Acetil Coenzima Carboxilasa Leptina: AMPK es activada, ACC es inhibida y caen los niveles de malonyl-CoA, incrementa la oxidación de ácidos grasos y reduce el contenido de lípidos de la célula. El contenido de lípidos reducido en el músculo citoesquelético permite la señalización insulina y el consumo de glucosa para retornar a niveles normales, reduciendo la resistencia a la insulina.
- 25. Papel de serin kinasas en stress oxidativo que induce resistencia a la insulina Hiperglicemia eleva los ácidos grasos libres, citoquinas, incrementa producción de ROS y stress oxidativo = activan serin treonin kinasa (IKKB) que fosforilan entre otras IRS y disminuye la estimulación de tirosin kinasa fosforilación incluyendo las moléculas corriente abajo lo cual lleva a una acción reducida de la insulina
- 26. La hiperglicemia causa elevados niveles intracelulares de glucosa en cells específicas. La inhibición de GAPPH suprime el metabolismo glicolítico causando diversos metabolitos glicoliticos dentro de las vías de sobre-utilización de la glucosa. Las cuatro vías resultantes generan stress oxidativo y promueven la progresión de complicaciones diabéticas AGE (productos finales de avanzada glicosilación)
- 27. Crecimiento del riñón diabético : hiperglicemia induce factores de crecimiento para proliferación temprana. TGF-B/p27 que media arrest de G1 de hiperplasia a hipertrofia y contribuye con ROS oxidativo de la célula. Suprime ODC (factor cto tubular aberrante) y causa crecimiento ECM y fibrosis (IGF) factor crecimiento de la insulina), ECM (matriz extracelular), Por qué hay proliferación y G1 arrest? High glucose treatment of a kidney mesangial cell line stimulates an early cell proliferative phase (24–48 h),and a later growth inhibitory phase (72–96 h) (Wolf et al.,1992)
- 28. Aterosclerosis en diabetes: Reacción no enzimática de glucosa y proteínas o lipo en pared arterial = Rx Maillard y o reacción de browning. Glicosilación reversible de proteínas con grupos amino reactivos circulantes o de pared. rearreglos más estables. Colageno pared celular del vaso Formación de productos glicosilados