
Recommandé
Contenu connexe
Tendances
Tendances (20)
Similaire à T20 IB Chemistry Organic
Similaire à T20 IB Chemistry Organic (20)
Plus de Robert Hughes
Dernier
Dernier (20)
T20 IB Chemistry Organic
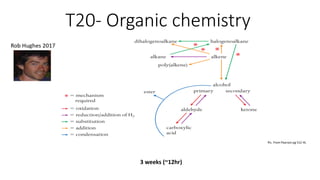
- 1. T20- Organic chemistry 3 weeks (~12hr) Pic. From Pearson pg 512 HL
- 2. 20.3- Sterioisomerism (Considering molecules in 3D space)
- 3. Lesson outline…. Lesson 1- Fundamentals of organic chemistry (T10). Lesson 2- Answering questions on each of the key families (T10). Lesson 3- Sterioisomerism- cis/trans and E/Z isomers. Lesson 4- Sterioisomerism – Optical isomers.
- 4. Lesson 1- Fundamentals of organic chemistry (T10). Level 4: recall what the rules are for naming organic compounds. Level 7: Explain why benzene is not susceptible to electrophilic addition. Level 5/6: Identify functional groups and use them to explain trends in boiling points.
- 6. Starter- A quick review of organic chemistry. Task 1- Rules for naming- see handout (note you only need to be able to name the following: alkanes, alkenes, halogenoalkanes, alcohols, ketones, aldehydes , carboxylic acids and esters and simple ethers). Task 2- See questions on fundamentals of organic chemistry from inthinking 1 &2.
- 7. Lesson 2- Answering questions on each of the key families (T10). Level 4: State what the reaction type is for key organic families. Level 7: Devise a synthetic pathway which links all organic families together. Level 5/6: Write simple equations to show the reactions of these key organic families.
- 8. Alkanes, alkenes, benzene and halogenoalkanes. Remember: • Electrophile- positive particle, attacks double bonds. • Nucleophile- negative particle, attacks polar carbon bond. • 3 reaction types: addition, substitution, oxidation. See questions worksheet from inthinking on each of these families.
- 9. Lesson 3- Stereoisomerism- cis/trans and E/Z isomers. Level 4: Construct 3-D models of a range of stereoisomers. Level 7: Explain why the physical properties of two isomers can be different. Level 5/6: Apply the Cahn-Ingold-Prelog priority system to naming E/Z stereoisomers.
- 10. Starter- Structural isomerism- isomers in 2D. Two organic molecules can have the same molecular formula but different structural formulas (atoms and functional groups attached in different ways). e.g. C2H6O Draw and name the two possible arrangements of this in 2D.
- 11. Stereoisomerism- isomers in 3D. From Pearson pg. 515 e.g. 1,2-dimethoxyethane
- 12. cis-trans isomerism • When a molecule contains double bonded carbons with two different groups attached OR a cyclic compound with the same situation, these can be arranged to give two different isomers:
- 13. Task- Molymod activity☺ Draw and make the following cis/trans isomeric pairs: cis- but-2-ene Vs. trans- but- 2-ene. cis- 1,2- dimethylcyclopropane Vs. trans-1,2- dimethlycyclopropane. cis- 1,3- dichlorocyclobutane Vs. trans- 1,3- dichlorocyclobutane. *Note: Notice how you cannot rotate the double bond/cyclised bond to switch between isomers as you would be able to do with single bonded conformational/structural isomers.
- 14. E/Z naming system • The E/Z naming system is more superior than the cis/trans system and so the cis/trans system is beginning to become obsolete. • The E/Z system can work for double bonded carbons which have more than 2 different groups between them: e.g. 1-bromo-2-methylbut-1-ene
- 15. The Cahn-Ingold-Prelog rules of priority. Q. So which isomer is represented here, Z or E?☺
- 16. Let’s practice applying those rules☺
- 17. The answers☺ (from inthinking)
- 18. Cis and trans fats….. https://www.youtube.com/watch?v=hE4xBd_7DCc
- 19. Lesson 4- Sterioisomerism – Optical isomers. Level 4: Use dashes and wedges to represent two enantiomers. Level 7: Explain how to separate a racemic mixture into its two enantiomers. Level 5/6: Discuss how scientists came up with the +/- or D/L naming system for optical isomers.
- 20. Stereoisomerism- isomers in 3D. From Pearson pg. 515
- 21. starter https://www.youtube.com/watch ?v=AnmWTruibMY B.Bad chiral compounds according to Walter White☺
- 22. Key terms Chiral carbon- A carbon which has four different atoms/groups attached to it. (a.k.a. asymmetric carbon or stereocentre). Optical isomers- Two isomers with a chiral carbon which are non-superimposable, mirror images of each other. They interreact in opposite ways with light (a.k.a. enantiomers). Racemic mixture/racemate- A mixture containing equal amounts of the two enantiomers, as a result it is optically inactive. Resolution: The separation of the two different enantiomers from a racemic mixture by reacting them with a single enantiomer of another compound. Diastereoisomerism- Can occur when a molecule has more than one chiral centre. It can result in two isomers which are not mirror images of each other. (common in sugars e.g. glucose is a diastereoisomer of galactose.
- 23. Optical isomerism • Carbon center with 4 different groups attached (‘chiral carbon’) • E.g. draw : 2-hydroxypropanoic acid (must use dash and wedge illustration for optical isomers) • 2 possible optical isomers will exist -2 ‘enantiomers.’
- 24. Optical isomers molymod activity • These 2 enantiomers are mirror images of each other. • They are non-superimposable. • They have the exact same physical and chemical properties eg. Mp, bp, density and polarity. May behave differently in biochemical systems.
- 25. Rotation of plane, polarized light • Two different enantiomers will rotate plane polarized light in opposite directions. Note: The concentrations of the solutions, the wavelength of light and the same path length must be kept the same for each measured value. Remember: A racemic mixture is an even mixture of the 2 different isomers which results in no activity in plane polarized light.
- 26. Diastereoimerism Diastereoisomerism- Can occur when a molecule has more than one chiral centre. It can result in two isomers which are not mirror images of each other. (common in sugars e.g. glucose is a diastereoisomer of galactose.
- 27. Optical isomerism in context • See handout on context for this form of isomerism & try the questions on the back ☺
- 29. Lesson outline…. Lesson 5- Understanding organic synthesis- The big picture. Lesson 6- Preparation of a halogenoalkane (P)
- 30. Lesson 5- Understanding organic synthesis- ‘The big picture.’ Level 4: State the definition for retro- synthesis. Level 7: Apply the retro-synthetic approach to the synthesis of an organic compound. Level 5/6: Deduce the correct synthetic route given starting reagents and a target product.
- 31. Starter- What is organic synthesis? Great 2min clip☺ https://www.youtube.com/watch?v=rh0Tn_oPS30
- 32. Pathways to know (*Practice memorising) * These mechanisms will be discussed in the next section. Picture from Pearson HL pg 512 Task- Try and add each reagent and any special conditions for each step. 5 types of reaction: Free- radical substitution S- Substitution (SN1/SN2) O- Oxidation. A- Addition (electrophilic) R- Reduction.
- 33. How good are you at identifying synthetic pathways?!
- 34. The synthesis of phenylamine Note: You need to know the mechanism for this reaction and we will discuss this in the next section.
- 35. Retro-synthesis: Working backwards. • The working backwards from a target molecule to generate precursor molecules for use in other reactions. • More efficient. • Can use computational modelling to identify what precursor molecules are contained within the target molecule. • Allows for the faster synthesis/discovery of useful precursor molecules. E.J. Corey
- 37. Worked example #2 This type of arrow is unique to retrosynthesis and means ‘can be made from’
- 38. Check for learning…. • See questions from inthinking on synthetic routes…
- 39. Lesson 6- Preparation of a halogenoalkane (P) Level 4: State the definition for a nucleophile. Level 7: Explain the purpose of each step that you carry out in this experiment. Level 5/6: Carry out a procedure to produce a halogenoalkane from an alcohol.
- 40. Aim: To synthesise a halogenoalkane from an alcohol by nucleophilic substitution. See practical method on conference and follow the steps carefully.
- 41. 20.1- Types of organic reactions.
- 42. Lesson outline…. Lesson 7- Substitution on halogenoalkanes (SN1 Vs. SN2). Lesson 8- Answering questions on SN1 and SN2 mechanisms. Lesson 9- Attacking pi bond electrons in alkenes. Lesson 10- Attacking pi bond electrons in benzene. Lesson 11- Oxidation and Reduction of organic compounds. Lesson 12- End of topic review☺
- 43. *Lesson 7- Substitution on halogenoalkanes (SN1 Vs. SN2). Level 4: State what the symbols of SN1 and SN2 mean. Level 7: Explain why tertiary iodoalkanes are the easiest to substitute on. Level 5/6: Compare and contrast the SN1 and SN2 mechanisms.
- 44. Starter-A closer look at reaction types. • 5 types of reaction: • Free- radical substitution • S- Substitution • O- Oxidation. • A- Addition. • R- Reduction.
- 45. Free radical substitution on Alkanes Key points • Alkanes are very slow to react with bromine water. • Homolytic/even splitting of the bond in the Br2 molecule (use fish hook arrows) • Free halogen radicals generated. • Substitute for a H atom on an alkane to form a halogenoalkane. • Very slow reaction. Mechanism *From SL core content.
- 46. Nucleophilic substitution on Halogenoalkanes (print out series of questions on this) 1° halogenoalkane SN2 mechanism 3° halogenoalkane SN1 mechanism Note: you do not need to know the mechanism for a 2° halogenoalkane as it can undergo a mixture of both.
- 47. Interpreting SN1 and SN2 mechanisms. 1. What functional group is being substituted for the Cl in both of these reactions? 2. Why is it considered a nucleophile? 3. What do you think the ‘SN’ stands for in these mechanism names? 4. Why do you think the OH- can attack from the back of the halogenoalkane when it’s primary and not when it’s tertiary? 5. If ‘Homolytic fission’ was represented by a fish hook arrow to show an even splitting of the electron pair in a bond, what do you think we call the bond breaking in these mechanisms? 6. The ‘2’ in SN2 denotes that this reaction rate has an overall order of 2 (‘bimolecular’). Try and write the rate equation for this mechanism. 7. The ‘1’ in SN1 denotes that this reaction rate has an overall order of 1 (‘unimolecular’). Try and write the rate equation for this mechanism.
- 48. A closer look at the SN2 mechanism. • Notice how the arrival of OH- group from the back of the molecule causes it to turn itself inside out/invert. • This is due to Lp-Bp replusion. • This mechanism is said to be ‘stereospecific- the 3D arrangement of the reactants dictates the 3D arrangement of the products.
- 49. A closer look at the SN1 mechanism. • The carbocation intermediate has a trigonal planar shape. • The OH- can now attack from any side (not stereospecific). • The blue arrows represent an inductive effect- the 3 methyl groups donate electron charge to try and stabalise the positive carbon atom (primary halogenoalkanes cannot do this).
- 50. Choosing the correct solvent for your NaOH. SN2 mechanism • An aprotic, polar solvent is chosen. e.g. propanone, ethanenitrile (CH3CN), ethoxyethane. No hydrogen bonding possible with any of the reactants. SN1 mechanism • A protic, polar solvent is chosen. e.g. water, ethanol. Hydrogen bonding is possible and the water molecules surround and stabilise the carbocation in the same way that they would any other ion in solution.
- 51. The easiest Halogen to substitute for R-Iodine • In both mechanisms we are trying to remove a halogen atom and replace it with OH-. • The C-X bond is quite polar and the polarity of this bond differs depending on the identity of the halogen (X). • Based on this concept alone, we would expect the highly polarised C-F bond to be the easiest to break. • In fact, if you look up the bond strength of each halogen to carbon in your databooks, you will find that the C-I bond is the weakest because of the sheer growing size of the halogen atom (steric hindrance).
- 53. Lesson 8- Answering Qs on SN1 and SN2 mechanisms. Level 4: reinforce the concept of the SN1 and SN2 mechanisms. Level 7: Explain how to determine, practically, which mechanism is faster. Level 5/6: Deduce the mechanism of a given reaction.
- 55. The answers!
- 56. More practice☺ See questions on nucleophilic substitution from inthinking.
- 57. Lesson 9- Attacking pi- bond electrons in alkenes. Level 4: State the 2 main reasons why alkenes are susceptible to electrophilic addition reactions. Level 7: Explain Markovnikov’s rule in terms of the inductive effect. Level 5/6: Deduce the mechanisms for the addition reactions on alkenes.
- 58. starter There are many electrophilic addition reactions which can occur with alkenes. The double bond always breaks and all of the reactant molecule is added. An electrophile is a species which loves electrons. They are positive ions or have a partial positive charge e.g. H+ e.g. HBr e.g. Br-Br *They are also Lewis acids as they accept a pair of electrons.
- 59. 2 things making alkenes so reactive….. 1. Sp2 hybridised carbons (e.g. in ethene) giving it a trigonal planar shape which is more open to attack than if it was tetrahedral (sp3) [Note: ethyne is sp hybridised, has a triple bond, linear shape which is even more attractive to electrophiles!] 2. The π bond between the unhybridized pz orbitals of the two carbons is above and below the plane of the bond axis. This makes the two electrons in it less associated/attracted to the carbon nuclei and so easier to break during addition reactions than the σ bond.
- 60. Ethene + bromine – Halogenation reaction. (try and complete this simple mechanism) + *demo reaction with bromine water. Initially: Step 1. (slow) Step 2. (fast)
- 61. Ethene + hydrogen bromide- Hydrohalogenation. Based on what we just showed for the previous mechanism, try and write the two step mechanism for this reaction: Step 1: (slow) Step 2: (fast) *Note: Just as Iodine was easier to remove from a halogenoalkane in the substitution reactions we studied, so too is H—I easier to heterolyticically split than HF for example.
- 62. Propene + hydrogen bromide (asymmetric addition- Markovnikov’s rule) Propene is an asymmetric molecule. How do we decide which carbon to add the H to and which to add the Br to?! “Zee rich get richer”
- 63. The H will attach to the carbon that is already bonded to the most hydrogens. Minor product. Major product. Highest positive inductive effect.
- 64. Check for learning…. • See 4 questions from inthinking☺
- 65. Lesson 10- Attacking pi- bond electrons in benzene. Level 4: Recall why benzene undergoes electrophilic substitution reactions and not addition. Level 7: Explain how to generate the NO2 + ion using nitric acid & sulfuric acid. Level 5/6: Deduce the mechanism for the nitration of benzene.
- 66. Starter- check for learning… From Pearson, pg 508.
- 68. Benzene in summary (from core) Fuse school- Great video☺: https://www.youtube.com/watch ?v=jMonOaN72wo
- 69. Explaining the extra stability of benzene (see model of benzene again) • Each Carbon forms 3 single bonds with bond angles of 120⁰ • Each carbon is sp2 hybridised. • This leaves one delocalized electron, per carbon, free to move around in a circle. These free electrons can move around the conjugated pz orbitals. • The ring is too stable, cannot break it by addition reactions! • Bond order of 9 e-/6 carbons= 1.5 • Undergo electrophilic substitution reactions instead e.g. with NO2 + or Cl-Cl (with instantaneous dipole)
- 70. Electrophilic substitution on benzene. *Try and write the same mechanism but using NO2 + as the electrophile…
- 71. How is NO2 + (nitronium) made? The special conditions for this reaction are: 50° C, conc. HNO3 and conc. H2SO4. The stronger sulfuric acid protonates the nitric acid: Note: This lost proton from sulfuric acid is regained when the NO2+ substitutes for the H+ on the benzene ring- thus the sulfuric acid is acting as a catalyst! Great video of this synthesis: https://www.youtube.com/watch?v=ovHFjtxo-D4
- 72. Useful molecules made from benzene… Small quantities of nitrobenzene are used in flavourings and perfume additives, but its use is limited due to high toxicity. It is more likely to be a precursor to another molecule:
- 73. Check for learning…. (Q3 from inthinking)
- 74. Lesson 11- Oxidation and Reduction reactions. Level 4: State the conditions for the reduction of carboxylic acids, aldehydes, ketones and nitrobenzene. Level 7: Explain when to choose LiAlH4 over NaBH4 as a reducing agent. Level 5/6: Deduce the mechanism for the reduction of nitrobenzene.
- 75. Starter (Q2. from inthinking)
- 76. The answers!
- 77. Oxidation of alcohols (from SL core) Ketone No reaction. H+/Cr2O7 2-H CH3
- 78. Reduction back to alcohols (LiAlH4 or NaBH4) Ketone No reaction. H+/Cr2O7 2-H CH3
- 79. Choosing whether to use LiAlH4 or NaBH4? • NaBH4 is safer but less reactive. Incapable of reducing carboxylic acids. • LiAlH4 is more reactive and will reduce carboxylic acids and ketones.
- 80. Reduction of nitrobenzene ‘phenylamine.’ From Pearson HL pg 510. Note: phenylamine is an important precursor molecule in the production of many pharmaceuticals, azo dyes and the vulcanization of rubber.
- 81. Check for learning… • See questions from inthinking on reduction reactions.
- 82. Lesson 12- Topic 20 review☺ See end of chapter questions from Pearson.
- 83. Pearson pg 511 See handout
- 84. - Nitrobenzene - phenylamine