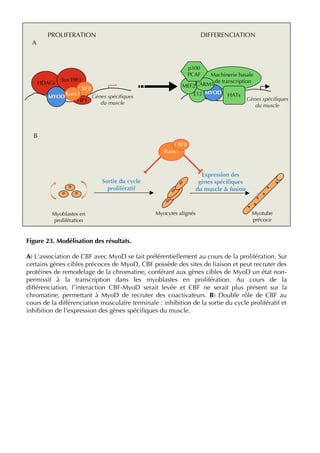
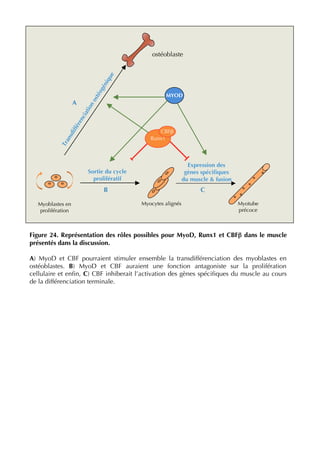
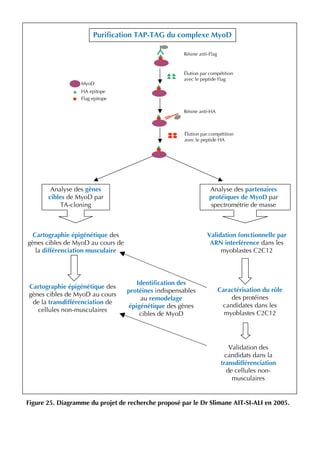
La thèse d'Ophélie Philipot, soutenue en octobre 2009 à l'Université Paris XI, explore la coopération entre le facteur de transcription cbf (runx1-cbfβ) et le facteur myogénique myod dans la régulation de l'équilibre entre prolifération et différenciation musculaire. Elle aborde en détail le rôle des différents types cellulaires constituant le muscle strié, ainsi que la dynamique des interactions entre myod et cbf dans les myoblastes et leur impact sur la différenciation musculaire. Le document met également en lumière l'importance des modifications post-traductionnelles et des voies de signalisation dans cette régulation complexe.














































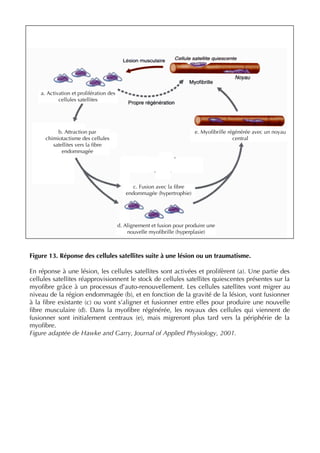















![37
pour les souris dont le gène JunB a été inactivé montre un phénotype étonnamment similaire
à celui des souris déficientes en CBFb. Par ailleurs, une étude du transcriptome dans les
cellules endothéliales de souris a révélé que CBFb était bien un gène cible de JunB. D’ailleurs
dans la moelle osseuse de souris déficientes en JunB, le niveau d’expression de CBFb est
diminué de 60% par rapport aux souris contrôles. Ceci montre que la régulation de
l’expression de CBFb est hautement dépendante de AP-1, en tout cas dans les cellules
endothéliales murines (Licht et al, 2006).
II.3 Relation Structure/ Fonction
II.3.1 La sous-unité protéique Runx1
Plusieurs domaines de la protéine Runx1 ont été conservés à travers l’évolution. On peut
notamment citer le domaine runt, les domaines d’autoinhibition, le domaine VWRPY, les
domaines de transactivation et la séquence de localisation nucléaire.
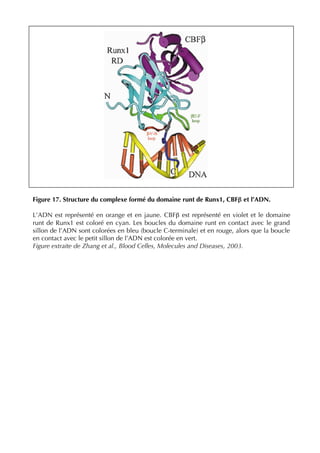
II.3.1.1 Le domaine runt de Runx1 : hétérodimérisation avec CBFβ et liaison à l’ADN
La famille de protéines à domaine runt est une famille de facteurs de transcription très
conservés, incluant Runt et Lozenge exprimés chez la drosophile, SpRunt chez les oursins,
Cs-Runt chez l’araignée (Cupiennius salei), Xam1 chez le xénope, Runx1, Runx2 et Runx3
chez les mammifères, RNT-1 (Caenorhabditis elegans). Le domaine runt (RD) est la signature
de cette famille de protéines. C’est un motif conservé de 128 acides aminés, responsable à la
fois pour la liaison à l’ADN de manière spécifique de séquence et de la dimérisation avec
CBFβ (Kagoshima et al, 1993). Le domaine runt de Runx1 interagit avec une région de 135
acides aminés de la protéine CBFβ. La stœchiométrie du complexe [Runx-CBFβ-ADN]
prédite et vérifiée est de 1:1:1. (Bravo et al, 2001; Ogawa et al, 1993a; Tang et al, 2000a;
Zhang et al, 2003). Des études de fixation de la protéine Runx1 montrent qu’elle a une
affinité réduite à l’ADN, par rapport au domaine runt seul, suggérant l’existence de
domaine(s) inhibiteur(s) hors du domaine runt. Cette inhibition de fixation à l’ADN est levée
par la liaison de CBFβ à Runx1, stimulant ainsi sa fixation à l’ADN d’au moins quarante fois.
Au sein du domaine runt, les régions respectivement impliquées dans l’interaction avec CBFβ
et la fixation à l’ADN sont distinctes et ne se chevauchent pas (Nagata & Werner, 2001)
(figure 17).
Comme il sera mentionné dans la section « II.3.1.2 Les domaines d’autoinhibition (NRHn,
NRDBn et NRHc », la dimérisation de CBFβ avec Runx1 peut être induite et stimulée par
d’autres facteurs de transcription, tel que Ets-1. Cependant, Runx1/CBFβ a un rôle dans des](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-65-320.jpg)





![40
identifiée en testant d’autres mutants de délétion de Runx1-P2. L’élément de transactivation
(TE) minimal s’étend ainsi des résidus 291 à 331 (TE1) et la région adjacente à TE1 qui
comprend les résidus 332 à 371 (appelée TE2) augmente significativement la transactivation
du mutant de Runx1-P2 (figure 18). Enfin, un autre TE a été identifié car le mutant de
délétion TE3 (résidus 243-191) possède, lui aussi une activité de transactivation (Kanno et al,
1998) (figure 18).
Il existe également une région d’activation dans la partie amino-terminale de Runx1 mettant
en jeu la partie adjacente au domaine runt et l’hélice α du runt domaine lui-même. C’est une
région acide de 26 résidus très conservée entre les trois protéines Runx de mammifères (Liu
et al, 2006) (figure 18).
II.3.1.5 Le signal de localisation nucléaire
Les protéines Runx contiennent un signal de localisation nucléaire (NLS) conservé dans la
partie C-terminale au domaine runt qui permet la localisation des protéines Runx dans le
noyau de la cellule. De plus, les protéines Runx possèdent également une séquence ciblant la
matrice nucléaire (NMTS) comprenant les résidus 324-353 chez Runx1-P2 murin (Kanno et
al, 1998) ou 351-381 chez son homologue Runx1-P2 humain (Zeng et al, 1997). L’association
du facteur CBF à la matrice nucléaire serait un pré-requis pour la transactivation par Runx1-
P2 (Zeng et al, 1998), d’ailleurs, la séquence NMTS de Runx1 comprend le domaine
d’activation (TE1 et TE2) (Kanno et al, 1998) (figure 18).
II.3.2 La sous-unité protéique CBFβ
La protéine CBFβ est une petite protéine de 22 kDa environ. Elle ne possède pas de
domaines particuliers : pas de NLS, pas de NES, pas de site de liaison à l’ADN. Le rôle le
plus important pour CBFβ est de lier le domaine runt et induire un changement de
conformation de Runx qui expose ainsi le domaine de liaison à l’ADN (figure 20).
La comparaison des constantes de dissociation [Runx-ADN] et [Runx-CBFβ-ADN] montre
que CBFβ augmente l’affinité de liaison à l’ADN de Runx. Le mécanisme de stabilisation de
la fixation de Runx à l’ADN par CBFβ n’est pas encore clairement défini, en revanche la
structure du dimère a été élucidée par le groupe de Bushweller (Huang et al, 1999; Tang et al,
2000a; Zhang et al, 2003). Ce groupe a également montré que les 141 premiers acides aminés
(1-141) suffisaient à CBFβ pour se lier au domaine runt avec la même efficacité que la
protéine complète (182 ou 187 résidus) (Zhang et al, 2003). Ce fragment N-terminal de 141
résidus est également capable de restaurer l’hématopoïèse terminale dans les cellules souches
embryonnaires déficientes en CBFb (Miller et al, 2001). Il est possible que ce domaine soit le](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-71-320.jpg)

















![The Core Binding Factor CBF Negatively Regulates Skeletal Muscle
Terminal Differentiation
PHILIPOT Ophelie and AIT-SI-ALI Slimane*
UMR7216 Epigénétique et Destin Cellulaire, Centre National de la Recherche Scientifique (CNRS), Université
Paris-Diderot, 35 rue Hélène Brion, 75013 Paris, France. Previous address: Institut André Lwoff, CNRS, FRE2944,
7 rue Guy Moquet, 94800 Villejuif; Université Paris-Sud, France
Tel: (33)-1-5727-8919, Fax: (33)-1-1-5727-8910
*: Corresponding author, email: slimane.aitsiali@univ-paris-diderot.fr,
Running title: CBF in muscle diffeentiation
ABSTRACT
Core Binding Factor or CBF is a transcription
factor composed of two subunits,
Runx1/AML-1 and CBF beta or CBFβ. CBF
was originally described as a regulator of
hematopoiesis. Here we show that CBF is
involved in the control of skeletal muscle
terminal differentiation. Indeed,
downregulation of either Runx1 or CBFβ
protein level accelerates cell cycle exit and
muscle terminal differentiation. Conversely,
overexpression of CBFβ in myoblasts slows
terminal differentiation. CBF interacts directly
with the master myogenic transcription factor
MyoD in proliferating but not in differentiating
myoblasts. The MyoD/CBF complex contains
several chromatin modifying enzymes that
inhibits MyoD activity, such as HDACs. When
overexpressed, CBFβ induced an inhibition of
histone H3 acetylation at MyoD target
promoters. Finally, we show a preferential
recruitment of Runx1 protein on MyoD target
genes in proliferating myoblasts. Taken
together, our data show a new role for Runx1
and CBFβ in the control of the
proliferation/differentiation in skeletal
myoblasts.
Keywords: Differentiation, proliferation,
muscle, MyoD, Runx1/CBFβ
INTRODUCTION
Runx1 (for Runt-related factor, also known as
AML1 for Acute Myeloid Leukemia 1,
CBFA2 or PEPB2aB) belongs to a family of
highly homologous heterodimeric transcription
factors named Core Binding Factors or CBF
(reviewed in: [1]). To be fully functional as a
transcription regulator, DNA binding subunit
Runx1 must dimerize with its cofactor CBF-
beta (CBFb, a non-DNA-binding subunit that
is expressed in a ubiquitous manner [2] Runx1
was originally identified at a breakpoint on
human chromosome 21 in the t(8;21)
translocation, known as the most common
target of chromosomal translocations in human
leukemia [3,4]. Genetic studies showed that
Runx1 is essential in the developing murine
embryo for definitive hematopoiesis of all
lineages [5,6].
There is now strong evidence that Runx
proteins are also important for differentiation
of multiple cell types, including osteoblasts
[7], neurons [8,9], hematopoietic cells of all
lineages [5,6,10] and skin epidermis and hair
follicle stem cells [11,12]. Runx1 is also
involved in promoting senescence in primary
mouse fibroblasts [13], and in cell cycle
regulation [14-16].
Runx proteins have the potential to either
activate or repress transcription in a context
dependent manner. Runx1 seems to promote
proliferation in progenitor cells, whereas in
differentiating cells it cooperates with tissue-
specific transcription factors to regulate tissue-
specific gene expression. For example, Runx1
cooperates with C/EBPα and C/EBPβ to
regulate hematopoiesis and osteogenesis,
respectively [17,18]. The dual role of Runx1 in
regulating proliferation and differentiation
could depend on differential interactions with
protein partners, specific for each stage of cell
development. The molecular mechanisms
underlying such a switch in Runx1 function
remain however to be deciphered.
Runx1 and CBFβ have also been linked to
skeletal muscle differentiation [19-21], and
prevention of muscle wasting [20]. In skeletal
muscle, proliferation and differentiation are](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-91-320.jpg)
![mutually exclusive. Indeed, skeletal muscle
terminal differentiation begins with an
irreversible withdrawal from the cell cycle,
followed by muscle-specific marker expression
[22]. Irreversible cell cycle exit involves a
definitive silencing of proliferation-associated
genes (reviewed in [23] and references
therein). Terminal muscle differentiation is
orchestrated by myogenic bHLH transcription
factors, such as MyoD and Myf5, two master
myogenic determination factors. MyoD is
expressed in proliferating myoblasts, but is
unable to activate its target genes even when
bound to their promoters [24,25]. MyoD
therefore has a repressive role at its target
genes prior to initiating chromatin remodeling
in differentiating cells [24,26,27]. In
proliferating myoblasts, MyoD is associated
with histone deacetylases (HDACs), the
histone methyltransferase Suv39h1 and
heterochromatin protein HP1, and might
actively inhibit expression of its target genes
by inducing a local repressive chromatin
structure [24,28,29].
Here we show that CBF associates with MyoD
only in proliferating myoblasts, and
knockdown of Runx1 or CBFβ accelerates cell
cycle exit and terminal differentiation.
Conversely, overexpression of CBF slows cell
cycle exit and delays muscle differentiation. In
proliferating myoblasts, the MyoD/CBF
complex contains several chromatin modifying
enzymes such as HDACs. In agreement with
this, when overexpressed, CBFβ maintains
histone H3 deacetylated on MyoD target
promoters even in differentiation conditions.
Finally, Runx1 is recruited to MyoD target
genes only in proliferating myoblasts, when
these genes are repressed. Altogether, our data
suggest that CBF transcription factor plays a
pivotal role as a negative regulator of skeletal
muscle terminal differentiation.
RESULTS
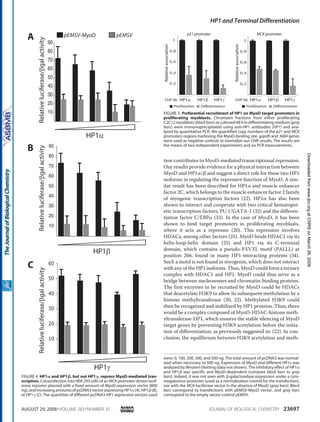
CBF subunits, Runx1 and CBFβ, interact
with MyoD in proliferating myoblasts
In an attempt to charaterize MyoD protein
partners, we carried out double-affinity
purification of HA-Flag MyoD stably
expressed in HeLa cells (see purification
scheme on Supplementary Figure 1). MyoD
protein complex composition was then
analyzed by mass spectrometry (MS) and
western blot (WB). MS analysis of the purified
protein complex revealed some already known
partners of MyoD (Supplementary Figure 2),
such as Id, Pbx1, PC4 and E12/E47, and new
partners that had never been described to
interact with MyoD. Indeed, MS analysis
unveiled CBFβ protein within MyoD complex
with a high number of peptides, covering
almost 20% of its protein mass and amino
acids content (Figure 1A). WB analyses
confirmed that result and showed that Runx1
also copurified with MyoD (Figure 1B).
We then performed a complementary
experiment by transfecting HA-tagged MyoD
and/or HA-tagged Runx1 into HeLa cells
stably expressing Flag-HA-CBFβ (ectopic,
eCBFβ). We showed that, indeed, MyoD co-
precipitated with eCBFβ (Figure 1C).
Moreover, the simultaneous co-transfection of
HA-tagged Runx1 resulted in its co-
precipitation with CBFβ and, more
importantly, increased the MyoD-CBFβ co-
precipitation (Figure 1C).
To further investigate the CBF/MyoD
interaction, we turned to myogenic cells: the
murine myolastic cell line C2C12. Both CBFβ
and Runx1 are expressed in the C2C12
myoblasts and their protein level do not vary
significantly during differentiation
(Supplementary Figure 3). We found that
MyoD and CBFβ co-precipitated preferentially
in proliferating compared to differentiating
C2C12 myoblasts (Figure 1D).
To assay whether MyoD has the ability to
interact directly with CBF, we performed GST
pull-down experiments, which showed that
GST-MyoD strongly interacts with Runx1
(Figure 1E, lane 4), but not with CBFβ (Figure
1E, lane 5). The interaction of MyoD with
Runx1 was specific; we did not detect any
Runx1 signal in the presence of GST protein
alone (Figure 1E, lane 8) nor any luciferase
signal with GST-MyoD (Figure 1E, lane 7).
Interestingly, GST-MyoD interacts with CBFβ
only in the presence of Runx1 (Figure 1E,
compare lanes 5 and 6), in agreement with our
previous findings (Figure 1C). These results
show that MyoD interacts directly with
heterodimeric transcription factor CBF, via the
Runx1 subunit.](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-92-320.jpg)

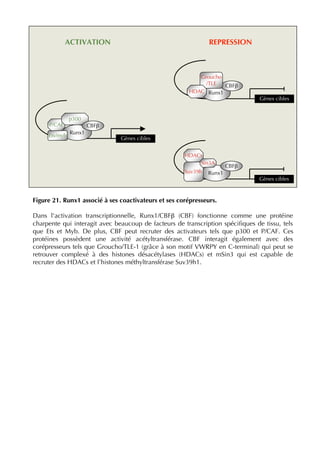
![transcriptional repression: the histone 3 lysine
9 (H3K9) methyltransferase Suv39h1, the
heterochromatin protein HP1β, and the histone
deacetylases HDACs 1, 2 and 3 (Figure 3B).
These proteins are already known partners of
Runx1 on the one hand [30], and repressors of
MyoD activity on the other hand [31-33].
Given the association of CBFβ with
chromatin-modifying enzymes, we studied the
chromatin status of three target gene promoters
of MyoD in differentiating C2C12-CBFβ using
chromatin immunoprecipitation (ChIP). Our
results showed that, in contrast to control cells
in which histone H3 acetylation (a mark
associated with transcription activation) on
myogenin, cyclin D3 and p21 promoters
increased in differentiation compared with
proliferation conditions, histone H3 acetylation
levels at these promoters did not vary in
C2C12-CBFβ cells (Figure 3C). This result is
an agreement with our previous findings
showing that CBF associates with chromatin
modifying enzymes, such as HDACs, Suv39h1
and HP1β, which are known to repress MyoD
activity in proliferating myoblasts. Thus, CBF
has an effect on the chromatin structure of
three early target genes of MyoD and
contibutes to maintain a repressive chromatin
state.
DISCUSSION
Here we show that CBF transcription factor is
expressed in proliferating myoblasts where it
interacts with MyoD. We found that
modulating the expression levels of either of
the CBF subunits, Runx1 or CBFβ, impaired
cell cycle exit and terminal myogenic
differentiation.
Runx1 or CBFβ downregulation in myoblasts
induced an accelerated cell cycle exit (cyclin
D1 protein disappears 24 hours earlier) in
differentiation conditions. In addition, Runx1
or CBFβ downregulation led to a an increase in
G1-phase cells, suggesting that CBF positively
regulates myoblasts proliferation. Conversely,
CBFβ overexpression delayed cyclin D1
disappearance even in differentiation
conditions (cyclin D1 is still detectable 72
hours after the induction of differentiation),
demonstrating a delayed cell cycle exit.
Consequently, entry into terminal
differentiation was delayed. In agreement with
this, our ChIP results showed that in
proliferating myoblasts, Runx1 is recruited to
repressd p21 and cyclin D3, which encode cell
cycle regulators. Together, these results
suggest that CBF regulates positively
proliferation, and negatively terminal
differentiation, of skeletal myoblasts. Thus,
CBF impacts on the
proliferation/differentiation switch in
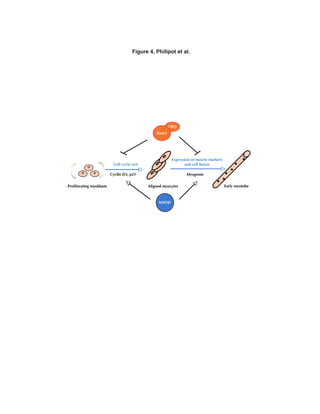
myoblasts (see model on Figure 4).
Our results revealed that such a role for CBF is
likely to be partly mediated through interaction
with MyoD. Indeed, we provide evidence that
CBF is recruited to early MyoD target genes in
proliferating myoblasts, where MyoD is
mainly associated with transcriptional
repressors [24]. This suggests that CBF may
serve for assembly of a transcription repression
complex at early MyoD target genes such as
p21 and cyclin D3. As for example, such a
mechanism could be involved in the repression
of the skeletal muscle acetylcholine receptor
gene, which contains a repressive E-box that
mediates its repression in proliferating
myoblasts [34]. In agreement with this, we
found that in proliferating myoblasts, CBF
associates with many chromatin modifying
enzymes, such as HDACs (1, 2 and 3),
Suv39h1, and HP1β, which are known to
repress MyoD activity in proliferating
myoblasts [24,31,35,36]; and already known to
interact with Runx1 [30,37,38]. We did not
succeed to show the concomitant presence of
Runx1 and MyoD on MyoD target genes,
given that it has reviously been demonstrated
that in proliferating conditions, a small fraction
of MyoD contributes to the repressive
remodeling of its target genes, prior
differentiation. Alternatively, Runx1 could
prevent the proper binding of MyoD and the
recruitment of the transcriptional machinery.
Notably, the displacement of Runx1 in
differentiating conditions is concomitant with a
strong binding of MyoD to its target promoters
(data not shown).
Our results showed that Runx1 was not located
on late target genes of MyoD, while it is
recruited into early muscle differentiation
genes. This suggests that Runx1 essentially
regulates cell cycle exit and early events of
skeletal muscle terminal differentiation.
Taken together, our results strongly suggest
that CBF could be recruited onto early MyoD](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-94-320.jpg)
![target genes to repress them in proliferating
myoblasts.
Interestingly, although MyoD is expressed
both in proliferating and differentiating cells,
we found that the interaction between MyoD
and CBF was lost in differentiating cells. In
proliferating myoblasts, a fraction of MyoD
could be recruited to its target genes where it
acts as a transcriptional repressor, until
differentiation starts [24]. We showed that
overexpression of CBFβ in myoblasts led to
stabilization of Runx1 that could more
efficiently repress MyoD transactivating
activity, which induces a delay in terminal
differentiation. In agreement with this, in
myoblasts overexpressing CBFβ, histone H3
acetylation (a mark of active transcription) on
MyoD target genes is delayed in differentiation
conditions.
In summary, we propose that CBF
transcription factor might participate in
recruiting chromatin modifying enzymes to
repress MyoD early target genes by locally
inducing a repressive chromatin structure. Our
data reveal a new critical role of CBF in the
regulation of the balance between proliferation
and differentiation in skeletal muscle cells.
They also demonstrate a new mechanism of
repression of differentiation genes in
proliferating myoblasts.
MATERIALS AND METHODS
Cell culture
C2C12 (CRL-1772, ATCC, USA) and HeLa S3
cells (CCL-2.2TM
, ATCC, USA) were cultured
under standard conditions. C2C12 cells and mouse
primary myoblasts (described in: [39]), were
cultured and differentiated as described in: [39].
Stable cell lines establishment and plasmid
construction
A HeLa cell line stably expressing MyoD was
established with a transgene encoding for full-
length MyoD; and HeLa and C2C12 cell lines
expressing CBFβ were established with a transgene
encoding for full-length CBFβ. The transgenes
were tagged with double-HA (Haemagglutinin) and
double-FLAG epitopes at the N-terminus as
described in [40].
Control cell lines transduced with the empty vector
were also established. Murine CBFβ cDNA (a kind
gift from Dr Nancy A. Speck) was amplified by
PCR with specific primers with protruding
restriction sites (fw-Pspx1:
CCGCTCGAGCCGCGCGTCGTCCCGGG, rev-Not1:
ATTCTATATGCGGCCGCTAACGAAGTTTGAGATC
ATCG, and subcloned into the XhoI-NotI sites in the
pREV retroviral vector after Pspx1 and Not1
digestion (Pspx1 is compatible with Xho cloning
site in the pRev vector) [40,41].
Protein complex purification
Flag-HA-MyoD complex purification from HeLa-
MyoD cells was performed as described in: [31]. 3
grams of C2C12-CBFβ cell pellet were used to
purify tagged CBFβ using a simple-affinity
purification method using Flag resin.
Preparation of nuclear extracts
Cells were scraped in a minimal volume of PBS and
centrifuged 2 min at 400 g. The pellet was
resuspended in: 20 mM Hepes pH 7, 0.15 mM
EDTA, 0.15 mM EGTA, 10 mM KCl, then lysed
by addition of NP-40 up to 4.5%. Nuclei were
immediately neutralized with addition sucrose
buffer (50 mM HEPES pH 7, 0.25 mM EDTA, 10
M KCl, 70% (m/v) sucrose). After centrifugation (5
min, 2000 g), nuclei were suspended in glycerol
buffer (10 mM HEPES pH 8, 0.1 mM EDTA, 100
mM NaCl, 25% glycerol) to remove any trace of
cytosolic components and centrifuged again. The
nuclei were then resuspended in sucrose buffer n°2
(20 mM Tris pH 7.65; 60 mM NaCl; 15 mM KCl;
0.34 M Sucrose) then lysed in a final concentration
of 250 mM NaCl using High Salt Buffer (20 mM
Tris pH 7.65; 0.2 mM EDTA; 25 % glycerol; 900
mM NaCl; 1.5 mM MgCl2). The lysates were
sonicated 3 times for 15s with the BioRuptor
(Diagenode, Liège, Belgium) on “High”, then
centrifuged 10 min at 13000 rpm to harvest the total
nuclear extracts (supernatants). Protein
concentration for each sample was estimated with
BCA kit (Perbio, Brebières, France).
Transient plasmid transfection and Flag-affinity
precipitation of Flag-HA-CBFβ
For plasmid transfection, 25 µg of pRcCMV-HA-
AML1 (kind gift of Dr I. Kitabayashi, Japan),
pCMV-HA-MyoD or pRC-CMV backbone were
transfected into HeLa-CBFβ cells, using calcium
phosphate pH 7.12, and Flag IPs was performed 24
h post-transfection (results presented on Figure 1C).
Each IP was performed with 1.5 mg of total nuclear
extracts and with 25 µL stock of ssDNA and BSA-
pre-blocked Agarose Flag M2 resin from Sigma. IP
was performed on wheel overnight at 4 °C. Resin
was then washed 5 times with TEGN buffer (20
mM Tris pH 7.65; 0.1 mM EDTA; 10 % glycerol;
150 mM NaCl; 0.5 % NP-40) and eluted by
competition with high-purity Flag peptide at a final
concentration of 0.2 mg/ml. The resin-free eluate
was retrieved using Clean-up Post reaction columns
(Sigma).](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-95-320.jpg)
![siRNA transfection
siRNAs were purchased from Sigma (Saint-Quentin
Fallavier, France) and were transfected using Hi-
Perfect reagent (Qiagen, Courtaboeuf, France)
according to the manufacturer recommendations.
We usually transfect 0.2 µmol of siRNA per 100
mm cell culture dish. CBFβ targeting siRNA
sequences used are: C1:
CCGGGAAUAUGUCGACUUA, and C2:
UAACUUAGGUGGCGGUGAU; Runx1 siRNA:
CUGUGAAUGCUUCUGAUUU; and the scrambled
siRNA: ACUUAACCGGCAUACCGGCTT.
Immunoprecipitation of endogenous proteins
For IP, we usually use 2 µg of antibodies, 10 µL
protein A/G Sepharose beads from Perbio and 1.2
mg of nuclear extracts from C2C12 cells, or 0.5 mg
from primary myoblasts. Elution was performed
with 40 µl of 0.1 M glycine pH 2.5, 15 min at 25
°C, the eluate was recovered using Spin cleaning-up
post-reaction column (Sigma). Acidity was
neutralized with Tris pH 8.0 before adding loading
buffer.
Western blotting
For western blotting, protein samples were resolved
on pre-cast NuPage 4-12% bis-Tris acrylamide
gradient SDS-PAGE gel (Invitrogen, Cergy-
Pontoise, France). Proteins were then transferred
onto nitrocellulose membrane during 1 h at 400 mA
in transfer buffer (25 mM Tris, 150 mM Glycine,
0.1 % SDS and 20 % methanol). Membranes are
blocked 1 hour in PBS-0.2 % Tween, 10 %
skimmed milk and incubated overnight at 4°C with
primary antibodies. Membranes were incubated
with the appropriate secondary antibodies coupled
to HRP and revealed using West Dura from Pierce
(Perbio, Brebières, France) and ChemiSmart 5000
system (Vilber Lourmat, Marne-La-Vallée, France).
Plasmids, GST fusions and GST pull-down
GST and GST-MyoD plasmid constructs were
expressed in Escherichia coli strain BL21 and
purified using glutathione-sepharose beads
according to the manufacturer (Sigma, Saint
Quentin-Fallavier, France). Purified proteins were
quantified by coomassie staining after SDS–PAGE
separation. In vitro transcription and translation
(TNT) of pcDNA3-AML1b, pcDNA3-CBFβ and
luciferase were performed with Riboprobe in vitro
transcription systems (Promega, Charbonnières,
France) in the presence of 35
S-labelled methionine.
Agarose beads coated with equal amounts of GST
or GST-MyoD (1 mg) were incubated with 10 µL
of radioactive TNT reaction in reaction buffer (50
mM Tris pH 7.6, 150 mM NaCl, 0.1% Triton)
during 2 h at 4°C. Beads were washed 5 times with
wash buffer (50 mM Tris pH 7.6, 300 mM NaCl,
0.5 % Triton 100), resuspended and proteins
resolved by SDS-PAGE gel and revealed by
autoradiography.
Immunofluorescence
Cells were cultured in Labtecks permanox (Falcon)
and fixed briefly with 4% formaldehyde in PBS.
Residual formaldehyde was neutralized with 0.1 M
Glycine pH 8.0, and washed with PBS. Cells were
permeabilized and blocked using 1 % BSA, 1 %
goat serum, 0.3 % Triton-X100 in PBS. Primary
and secondary antibodies were diluted in the
permeabilizing/blocking solution and were washed
with 0.3% Triton-X100 in PBS. Nuclei are stained
with DAPI and the glass lid is fixed using an anti-
fading polymerizing media from DakoCytomation
(Dako, Trappes, France).
Antibodies
The C-20 anti-MyoD, M-225 anti-myogenin, C-20
anti-Myf5, FL-182 anti-CBFβ normal rabbit IgG
antibodies were purchased from Santa Cruz Biotech
(Santa Cruz, CA, USA). Rabbit polyclonal anti-
Suv39h1 antibody (ref. 07-550), and rabbit anti-
acetyl histone H3 (ref. 06-599) were obtained from
Upstate Biotech (Lake Placid, NY, USA). Anti-
HP1β (1MOD1A9AS) was from Euromedex
(Souffelweyersheim, France). Rabbit polyclonal
anti-MCK antibody was developed by Dr H. Ito
[42]. Anti-Flag and anti-α-tubulin antibodies were
purchased from Sigma (Saint-Quentin Fallavier,
France). Rat anti-HA antibody was purchased from
Roche (Meylan, France). Mouse anti-AML1
antibody (MAB10062) was purchased from
Millipore (Saint Quentin en Yvelines, France) and
mouse anti-HDAC3 antibody (611125) from BD
Biosciences (Le Pont de Claix, France). Goat anti-
rat IgG Alexa-488-conjugated, anti-rabbit IgG
Alexa-488 were from Invitrogen (Cergy-Pontoise,
France) and anti-mouse IgG TRITC (T7657) were
from Sigma (Saint-Quentin Fallavier, France).
FACS analysis
C2C12 were transfected with the siRNAs as
indicated in the Material and Method section. 48
hours post-transfection, cells were washed with
PBS, then scraped in 500 µL of PBS. Cells were
kept on ice while 4,5 mL of ethanol 70% were
added. Then cells are kept at least overnight at -
20°C. Propidium iodide (PI) staining proceeds as
follows: cells are centrifuged and the pellet is
washed with PBS. Cells are then centrifuged and
resuspended in 2mL PI solution (PI, RNase, Triton
0,1%) 30 minutes and kept in the dark. Cells are
homogeneized by vortexing before analysis. We
worked on a Beckman and Coulter FACS apparatus
and we counted at least 3000 events for each
condition.
Chromatin immunoprecipitation (ChIP)](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-96-320.jpg)
![ChIP protocol and primers have been described in:
[39]. The yet unpublished primers used are:
Myogenin fw: GAATCACATGTAATCCACGGA, rev:
ACGCCAACTGCTGGGTGCCA. Cyclin D3 fw:
CTGCTTGCCTCTGTCTTCA; rev:
GACCCATGTCAGATGACTC. 36B4 fw:
ATGTGCAGCTGATAAAGACTGG; rev:
CTGTGATGTCGAGCACTTCAG.
ACKNOWLEGEMENTS
We thank P. Robin, C. Pellentz, L. Fritsch, A.
Polesskaya, and A. Marchand for technical
help and critical reading of the manuscript. We
thank F. Ferri and C. Guillemin for technical
help. We thank C. Francastel, J. Weitzman, F.
Hubé, C. Rougeulle, V. Mezger and P-A.
Defossez for critical reading of the manuscript.
The authors warmly thank Drs N. Speck, G.
Mouchiroud, I. Kitabayashi, H. Ito, and L.
Delva for sharing reagents. This work was
supported by the Association Française contre
les Myopathies (AFM); the Fondation
Bettencourt-Schueller; the Ligue Nationale
contre le Cancer; the Association pour la
Recherche sur le Cancer (ARC), the CNRS;
and the Université Paris-Sud. PO was recipient
of fellowships from the Ministère de la
Recherche and the ARC.
CONFLICT OF INTEREST
None of the authors has any commercial
affiliations, consultancies, or equity interests,
nor patent-licensing arrangements that could
be considered to pose a conflict of interest
regarding the submitted manuscript.
REFERENCES
1. Mikhail FM, Sinha KK, Saunthararajah Y,
Nucifora G (2006) Normal and
transforming functions of RUNX1: a
perspective. J Cell Physiol 207: 582-593.
2. Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL,
et al. (1996) The CBFbeta subunit is
essential for CBFalpha2 (AML1) function
in vivo. Cell 87: 697-708.
3. Miyoshi H, Shimizu K, Kozu T, Maseki N,
Kaneko Y, et al. (1991) t(8;21)
breakpoints on chromosome 21 in acute
myeloid leukemia are clustered within a
limited region of a single gene, AML1.
Proc Natl Acad Sci U S A 88: 10431-
10434.
4. Golub TR, Barker GF, Bohlander SK, Hiebert
SW, Ward DC, et al. (1995) Fusion of the
TEL gene on 12p13 to the AML1 gene on
21q22 in acute lymphoblastic leukemia.
Proc Natl Acad Sci U S A 92: 4917-4921.
5. Wang Q, Stacy T, Binder M, Marin-Padilla M,
Sharpe AH, et al. (1996) Disruption of the
Cbfa2 gene causes necrosis and
hemorrhaging in the central nervous
system and blocks definitive
hematopoiesis. Proc Natl Acad Sci U S A
93: 3444-3449.
6. Okuda T, van Deursen J, Hiebert SW, Grosveld
G, Downing JR (1996) AML1, the target
of multiple chromosomal translocations in
human leukemia, is essential for normal
fetal liver hematopoiesis. Cell 84: 321-
330.
7. Komori T, Kishimoto T (1998) Cbfa1 in bone
development. Curr Opin Genet Dev 8:
494-499.
8. Chen CL, Broom DC, Liu Y, de Nooij JC, Li Z,
et al. (2006) Runx1 determines nociceptive
sensory neuron phenotype and is required
for thermal and neuropathic pain. Neuron
49: 365-377.
9. Theriault FM, Nuthall HN, Dong Z, Lo R,
Barnabe-Heider F, et al. (2005) Role for
Runx1 in the proliferation and neuronal
differentiation of selected progenitor cells
in the mammalian nervous system. J
Neurosci 25: 2050-2061.
10. Ichikawa M, Asai T, Saito T, Seo S, Yamazaki
I, et al. (2004) AML-1 is required for
megakaryocytic maturation and
lymphocytic differentiation, but not for
maintenance of hematopoietic stem cells
in adult hematopoiesis. Nat Med 10: 299-
304.
11. Raveh E, Cohen S, Levanon D, Negreanu V,
Groner Y, et al. (2006) Dynamic
expression of Runx1 in skin affects hair
structure. Mech Dev 123: 842-850.
12. Osorio KM, Lee SE, McDermitt DJ, Waghmare
SK, Zhang YV, et al. (2008) Runx1
modulates developmental, but not injury-
driven, hair follicle stem cell activation.
Development 135: 1059-1068.
13. Wotton SF, Blyth K, Kilbey A, Jenkins A,
Terry A, et al. (2004) RUNX1
transformation of primary embryonic
fibroblasts is revealed in the absence of
p53. Oncogene 23: 5476-5486.
14. Britos-Bray M, Ramirez M, Cao W, Wang X,
Liu PP, et al. (1998) CBFbeta-SMMHC,
expressed in M4eo acute myeloid
leukemia, reduces p53 induction and slows
apoptosis in hematopoietic cells exposed
to DNA-damaging agents. Blood 92:
4344-4352.
15. Cao W, Adya N, Britos-Bray M, Liu PP,
Friedman AD (1998) The core binding
factor (CBF) alpha interaction domain and](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-97-320.jpg)












































![107
between heterologous helix-loop-helix proteins generate complexes that bind specifically
to a common DNA sequence. Cell 58(3): 537-544
Mussini I, Favaro G, Carraro U (1987) Maturation, dystrophic changes and the continuous
production of fibers in skeletal muscle regenerating in the absence of nerve. J Neuropathol
Exp Neurol 46(3): 315-331
Nagata T, Werner MH (2001) Functional mutagenesis of AML1/RUNX1 and PEBP2 beta/CBF
beta define distinct, non-overlapping sites for DNA recognition and heterodimerization
by the Runt domain. J Mol Biol 308(2): 191-203
Naidu PS, Ludolph DC, To RQ, Hinterberger TJ, Konieczny SF (1995) Myogenin and MEF2
function synergistically to activate the MRF4 promoter during myogenesis. Mol Cell Biol
15(5): 2707-2718
Nam S, Jin YH, Li QL, Lee KY, Jeong GB, Ito Y, Lee J, Bae SC (2002) Expression pattern,
regulation, and biological role of runt domain transcription factor, run, in Caenorhabditis
elegans. Mol Cell Biol 22(2): 547-554
Nguyen DX, Baglia LA, Huang SM, Baker CM, McCance DJ (2004) Acetylation regulates the
differentiation-specific functions of the retinoblastoma protein. Embo J 23(7): 1609-1618
Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T
(1997) Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene
encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer
binding protein 2/core binding factor. Proc Natl Acad Sci U S A 94(11): 5697-5702
Nimmo R, Antebi A, Woollard A (2005) mab-2 encodes RNT-1, a C. elegans Runx homologue
essential for controlling cell proliferation in a stem cell-like developmental lineage.
Development 132(22): 5043 - 5054
Nishimura M, Fukushima-Nakase Y, Fujita Y, Nakao M, Toda S, Kitamura N, Abe T, Okuda T
(2004) VWRPY motif-dependent and -independent roles of AML1/Runx1 transcription
factor in murine hematopoietic development. Blood 103: 562 - 570
Nishimura R, Hata K, Harris SE, Ikeda F, Yoneda T (2002) Core-binding factor alpha 1 (Cbfa1)
induces osteoblastic differentiation of C2C12 cells without interactions with Smad1 and
Smad5. Bone 31(2): 303-312
Norton JD, Deed RW, Craggs G, Sablitzky F (1998) Id helix-loop-helix proteins in cell growth
and differentiation. Trends Cell Biol 8(2): 58-65
Novitch BG, Mulligan GJ, Jacks T, Lassar AB (1996) Skeletal muscle cells lacking the
retinoblastoma protein display defects in muscle gene expression and accumulate in S and
G2 phases of the cell cycle. J Cell Biol 135(2): 441-456
Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB (1999) pRb is required for MEF2-
dependent gene expression as well as cell-cycle arrest during skeletal muscle
differentiation. Curr Biol 9(9): 449-459
Nurse P (1994) Ordering S phase and M phase in the cell cycle. Cell 79(4): 547-550
Ogawa E, Inuzuka M, Maruyama M, Satake M, Naito-Fujimoto M, Ito Y, Shigesada K (1993a)
Molecular Cloning and Characterization of PEBP2[beta], the Heterodimeric Partner of a
Novel Drosophila runt-Related DNA Binding Protein PEBP2[alpha]. Virology 194(1):
314-331
Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y (1993b)
PEBP2/PEA2 represents a family of transcription factors homologous to the products of](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-146-320.jpg)




![112
Strom DK, Nip J, Westendorf JJ, Linggi B, Lutterbach B, Downing JR, Lenny N, Hiebert SW
(2000) Expression of the AML-1 oncogene shortens the G(1) phase of the cell cycle. J
Biol Chem 275(5): 3438 - 3445
Sullivan J, Sher D, Eisenstein M, Shigesada K, Reitzel A, Marlow H, Levanon D, Groner Y,
Finnerty J, Gat U (2008) The evolutionary origin of the Runx/CBFbeta transcription
factors - Studies of the most basal metazoans. BMC Evolutionary Biology 8(1): 228
Sun L, Trausch-Azar JS, Ciechanover A, Schwartz AL (2005) Ubiquitin-proteasome-mediated
degradation, intracellular localization, and protein synthesis of MyoD and Id1 during
muscle differentiation. J Biol Chem 280(28): 26448-26456
Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M (1997) Redefining the Genetic Hierarchies
Controlling Skeletal Myogenesis: Pax-3 and Myf-5 Act Upstream of MyoD. Cell 89(1):
127-138
Tanaka T, Kurokawa M, Ueki K, Tanaka K, Imai Y, Mitani K, Okazaki K, Sagata N, Yazaki Y,
Shibata Y, Kadowaki T, Hirai H (1996) The extracellular signal-regulated kinase pathway
phosphorylates AML1, an acute myeloid leukemia gene product, and potentially regulates
its transactivation ability. Mol Cell Biol 16(7): 3967-3979
Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Nishida J, Shibata Y, Yazaki Y, Hirai H
(1995) An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell
differentiation and transcriptional activation antagonistically by two alternative spliced
forms. Embo J 14(2): 341-350
Tanaka Y, Watanabe T, Chiba N, Niki M, Kuroiwa Y, Nishihira T, Satomi S, Ito Y, Satake M
(1997) The protooncogene product, PEBP2beta/CBFbeta, is mainly located in the
cytoplasm and has an affinity with cytoskeletal structures. Oncogene 15(6): 677-683
Tang Y-Y, Crute BE, Kelley JJ, Huang X, Yan J, Shi J, Hartman KL, Laue TM, Speck NA,
Bushweller JH (2000a) Biophysical characterization of interactions between the core
binding factor [alpha] and [beta] subunits and DNA. FEBS Letters 470(2): 167-172
Tang Y-Y, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, Speck NA, Bushweller JH (2000b)
Energetic and Functional Contribution of Residues in the Core Binding Factor beta
(CBFbeta ) Subunit to Heterodimerization with CBFalpha. J Biol Chem 275(50): 39579-
39588
Taya Y (1997) RB kinases and RB-binding proteins: new points of view. Trends Biochem Sci 22(1):
14-17
Thayer MJ, Tapscott SJ, Davis RL, Wright WE, Lassar AB, Weintraub H (1989) Positive
autoregulation of the myogenic determination gene MyoD1. Cell 58(2): 241-248
Theriault FM, Nuthall HN, Dong Z, Lo R, Barnabe-Heider F, Miller FD, Stifani S (2005) Role
for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in
the mammalian nervous system. J Neurosci 25(8): 2050-2061
Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC (1999) The
nucleosomal response associated with immediate-early gene induction is mediated via
alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase.
EMBO J 18(17): 4779-4793
Tintignac LA, Leibovitch MP, Kitzmann M, Fernandez A, Ducommun B, Meijer L, Leibovitch
SA (2000) Cyclin E-cdk2 phosphorylation promotes late G1-phase degradation of MyoD
in muscle cells. Exp Cell Res 259(1): 300-307](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-151-320.jpg)

![114
Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ (2005) Runx1 prevents
wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 19(14):
1715-1722
Wasserman WW, Fickett JW (1998) Identification of regulatory regions which confer muscle-
specific gene expression. J Mol Biol 278(1): 167-181
Wee H-J, Voon DC-C, Bae S-C, Ito Y (2008) PEBP2{beta}/CBF{beta}-dependent
phosphorylation of RUNX1 and p300 by HIPK2: Implications for leukemogenesis. Blood:
blood-2008-2001-134122
Wei L, Jamonnak N, Choy J, Wang Z, Zheng W (2008) Differential binding modes of the
bromodomains of CREB-binding protein (CBP) and p300 with acetylated MyoD.
Biochemical and Biophysical Research Communications 368(2): 279-284
Weintraub H, Davis R, Lockshon D, Lassar A (1990) MyoD binds cooperatively to two sites in a
target enhancer sequence: occupancy of two sites is required for activation. Proc Natl Acad
Sci U S A 87(15): 5623-5627
Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991)
Muscle-specific transcriptional activation by MyoD. Genes Dev 5(8): 1377-1386
Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD (1989)
Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines
by forced expression of MyoD. Proc Natl Acad Sci U S A 86(14): 5434-5438
Wong MW, Pisegna M, Lu MF, Leibham D, Perry M (1994) Activation of Xenopus MyoD
transcription by members of the MEF2 protein family. Dev Biol 166(2): 683-695
Wright WE, Sassoon DA, Lin VK (1989) Myogenin, a factor regulating myogenesis, has a
domain homologous to MyoD. Cell 56(4): 607-617
Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL
(2000) p38 and extracellular signal-regulated kinases regulate the myogenic program at
multiple steps. Mol Cell Biol 20(11): 3951-3964
Xia D, Zhang Y, Huang X, Sun Y, Zhang H (2007) The C. elegans CBF[beta] homolog, BRO-1,
regulates the proliferation, differentiation and specification of the stem cell-like seam cell
lineages. Developmental Biology 309(2): 259-272
Xie T, Spradling AC (1998) decapentaplegic is essential for the maintenance and division of
germline stem cells in the Drosophila ovary. Cell 94(2): 251-260
Xu Q, Yu L, Liu L, Cheung CF, Li X, Yee SP, Yang XJ, Wu Z (2002) p38 Mitogen-activated
protein kinase-, calcium-calmodulin-dependent protein kinase-, and calcineurin-mediated
signaling pathways transcriptionally regulate myogenin expression. Mol Biol Cell 13(6):
1940-1952
Yablonka-Reuveni Z, Paterson BM (2001) MyoD and myogenin expression patterns in cultures
of fetal and adult chicken myoblasts. J Histochem Cytochem 49(4): 455-462
Yaffe D, Saxel O (1977) Serial passaging and differentiation of myogenic cells isolated from
dystrophic mouse muscle. Nature 270(5639): 725-727
Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-
Bellan A, Ait-Si-Ali S (2008) Differential cooperation between heterochromatin protein
HP1 isoforms and MyoD in myoblasts. J Biol Chem: M802647200
Yamaguchi Y, Kurokawa M, Imai Y, Izutsu K, Asai T, Ichikawa M, Yamamoto G, Nitta E,
Yamagata T, Sasaki K, Mitani K, Ogawa S, Chiba S, Hirai H (2004) AML1 Is Functionally](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-153-320.jpg)




![Author Proof
Review
10.1517/14728222.10.6.xxx © 2006 Informa UK Ltd ISSN 1472-8222 1
General
Chromatin modification and
muscle differentiation
Hakima Yahi*, Ophélie Philipot*, Valentina Guasconi, Lauriane Fritsch
& Slimane Ait-Si-Ali†
†Institut André Lwoff, Laboratoire ‘Epigénétique et Cancer’, FRE 2944, CNRS, 7 rue Guy Moquet,
94800 Villejuif, France. *These authors contibuted equally to this manuscript.
Skeletal muscle differentiation is a multistep process, which begins with the
commitment of multi-potent mesodermal precursor cells to the muscle fate.
These committed cells, the myoblasts, then differentiate and fuse into multi-
nucleated myotubes. The final step of muscle differentiation is the maturation
of differentiated myotubes into myofibres. Skeletal muscle development
requires the coordinated expression of various transcription factors like the
members of the myocyte enhancer binding-factor 2 family and the muscle
regulatory factors. These transcription factors, in collaboration with
chromatin-remodelling complexes, act in specific combinations and within
complex transcriptional regulatory networks to achieve skeletal myogenesis.
Additional factors involved in the epigenetic regulation of this process con-
tinue to be discovered. In this review, the authors discuss the recent discoveries
in the epigenetic regulation of myogenesis. They also summarise the role of
chromatin-modifying enzymes regulating muscle gene expression. These dif-
ferent factors are often involved in multiple steps of muscle differentiation and
have redundant activities. Altogether, the recent findings have allowed a
better understanding of myogenesis and have raised new hopes for the
pharmacological development of new therapies aimed at muscle degeneration
diseases, such as myotonic dystrophy or Duchenne muscular dystrophy.
Keywords: chromatin remodelling, differentiation, skeletal muscle
Expert Opin. Ther. Targets (2006) 10(6):xxx-xxx
1. Introduction
Skeletal muscles derive from muscle precursor cells (MPCs) that engage in a
multistep differentiation programme in which mesoderm progenitors become com-
mitted to a skeletal muscle fate, then proliferate and migrate to specific locations,
and finally differentiate into multinucleated myotubes to form myofibres [1].
Among the different factors expressed in the embryo structures, members of
the Wnts family and Sonic Hedgehog (Shh) factor are positive regulators of myo-
genesis [2-7]. These factors induce the production of Myf5 and MyoD, the determi-
nation factors of myogenesis. Many other regulatory factors involved in these
different steps have been identified, including the early markers Pax3, c-Met, the
members of the MEF2 family and the muscle regulatory factors (MRFs): MyoD,
myogenin, Myf5, and MRF4 (Table 1). The MEF2 and MRFs transcription factors
are the key regulators of the latest steps of myogenesis. The ectopic expression of an
individual MRF in a variety of non-muscle cells results in the activation of the skel-
etal muscle differentiation programme [8,9]. These transcription factors, in collabo-
ration with chromatin remodelling complexes, act in specific combinations and
within complex transcriptional regulatory networks to achieve skeletal myogenesis.
1. Introduction
2. The specification factors of
embryonic myogenesis
3. Transcriptional regulation of
muscle differentiation
4. Chromatin remodelling
enzymes in proliferating
myoblasts
5. Chromatin remodelling
enzymes in differentiating
myotubes
6. Insights from muscle
regeneration studies
7. Pharmacological triggering of
the transcription regulators of
skeletal muscle differentiation
8. Conclusion
9. Expert opinion](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-158-320.jpg)
![Author Proof
Chromatin modification and muscle differentiation
2 Expert Opin. Ther. Targets (2006) 10(6)
2. The specification factors of embryonic
myogenesis
In the embryo, a number of factors are implicated in the sur-
vival, delamination and migration of the MPCs from the
dermomyotome to sites of muscle formation in the body and
limbs (Figure 1). Several homeodomain transcription factors are
involved. Thus, Pax3 and Pax7 mark cells in the dermomyo-
tome and in newly forming muscle masses, such as the myo-
tome in the mature somite. Pax3 is essential for the formation
and migration of the MPCs. Pax3 gene acts upstream of the
myogenic programme and is necessary for MyoD activation in
the absence of Myf-5 [10,11]. Most of the functions of Pax3 can
be replaced by its paralogue Pax7 [12]. Msx1 is a homeodo-
main protein that can inhibit myogenesis, with an antagonist
effect of Pax3 and the MRFs. The Six family (homeobox pro-
teins) are transcription factors with notably Six1 and Six4 that
are expressed in MPCs and can bind the MEF-3 element
located in myogenin promoter. Lbx1 is also a homeobox pro-
tein necessary for a correct migration of the MPCs [13].
Finally, the proto-oncogene c-met is a marker of the MPCs
delaminating and migrating from the lateral dermomyotome.
The c-met gene encodes a tyrosine kinase receptor and its lig-
and is the hepatocyte growth factor, also called scatter factor
because it delineates the migratory route of MPCs. During
migration, the MPCs express all these specification factors,
but do not yet express the determination factors, such as
MyoD and Myf-5 (Figure 1).
3. Transcriptional regulation of muscle
differentiation
The development of skeletal muscle is an ordered multistep
process requiring the sequential activation of essentially two
groups of myogenic transcription factors (Figures 1 and 2,
Table 1). The first group is composed of the
basic helix-loop-helix (bHLH) transcription factors family. The
second group includes the MEF2 family of transcription factors.
3.1 The myogenic bHLH transcription factors family
The myogenic bHLH family comprises MyoD, Myf5, myo-
genin and MRF4, which belong to the Class II of the tis-
sue-specific bHLH transcription factors family. Targeted gene
disruption of any of the four bHLH family members, MyoD,
Myf5, myogenin and Mrf4, in mice has shown that they play
central roles in myogenesis [14-16]. Progenitor cells remain
multi-potent in the absence of these factors, they do not
locate correctly to sites of myogenesis and adopt other cell
fates [17,18]. Moreover, in the absence of MyoD and Myf5,
myogenin and MRF4 are not observed [19-21]. On the con-
trary, it has been shown that mice lacking myogenin or MRF4
still develop skeletal muscle, although double knockout mice
totally lack skeletal muscle fibres and myoblasts [19-21]
(for review see [16]).
Further experiments have pointed out the compensatory
roles of Myf5 and MyoD. Inactivation of Myf5 gene results
in delayed myotome formation and aberrant migration of
Figure 1. Sequential expression of major markers of skeletal muscle differentiation and illustration of the different stages of
myogenesis, from the somitic cells to mature myofibres. The committed cells in the somite are characterised by the expression of
Pax3, which will further lead to the migration of the MPCs, along with c-Met. The expression of MyoD and Myf5 in the MPCs leads to
their determination into proliferating myoblasts. The concomitant expression of MyoD, Myf5, Mfr4 and the MEF2 factors, will induce
these myoblasts to exit from the cell cycle and to enter terminal differentiation. Late-stage markers of differentiation, such as MCK and
MHC, are expressed in the multinucleated myotubes. The latter finally fuse to form mature and functional myofibres.
MCK: Muscle creatine kinase; MEF: Myocyte enhancer factor; MHC: Myosin heavy chain; MPC: Muscle precursor cell; MRF: Muscle regulator factory; MyoD: Myogenic
determination.
Maturation
Committed myoblasts at the
site of muscle formation
Myotubes, at site of muscle
formation. Permanent cell
cycle exit
Mature myofibers
Uncommitted
cells of the
somite
DifferentiationDetermination
Terminally differentiatedProliferative
Specification
Muscle cell precursors
Pax3, Pax7
Wnt, Shh
Six1, Six4
c-met, lbx1, msx1
Myf5, MyoD, MRF4
MEF2
MyoD, MRF4,
Myogenin
MCK, MHC
Troponin1
AchR
MEF2](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-159-320.jpg)
![Author Proof
Yahi, Philipot, Guasconi, Fritsch & Ait-Si-Ali
Expert Opin. Ther. Targets (2006) 10(6) 3
muscle precursors into the adjacent tissues, dermis and
sclerotome, where they acquire non-muscle fates [22]. This
early defect in myogenesis is, however, compensated by the
later expression of MyoD, leading to essentially normal
muscles at birth. Similarly, disruption of the MyoD gene,
which is normally expressed two days after Myf5, does not
alter muscle formation. Indeed, homozygous MyoD-/- mice
are normal, fertile and do not show abnormal skeletal mus-
cles [23]. To understand this compensatory role, analysis of
the mRNA of newborn mice shows an increased level of
Myf5 expression, both in homozygous and heterozygous
MyoD+/- mice. This suggests that Myf5 replaces MyoD in
these mice and, thus, there is functional overlap between
these two genes.
MyoD positively auto-regulates its own expression, and
activates that of myogenin (Figure 2). Myogenin is expressed
in nonproliferating myoblasts as they enter the differentiation
pathway. Myogenin seems to be implicated in later steps of
muscle cell differentiation and triggers differentiation with
activation of muscle specific genes, such as muscle creatine
kinase (MCK) and myosin heavy chain (MHC).
Both MyoD and myogenin are expressed during skeletal
muscle differentiation and it is likely that, together, they reg-
ulate the late expression of MRF4, which contributes to
fibres’ maturation and to the maintenance of their differen-
tiated state (Figure 2). But Mrf4 is not only playing a role in
the late stages of muscle differentiation. Indeed, it was
recently shown that Mrf4 is expressed in MPCs, such as
MyoD and Myf5 [11,24,25]. Actually, Myf5 and MRF4 act
upstream of MyoD to direct embryonic multi-potent cells
into the myogenic lineage. Moreover, Myf5/MyoD double
null mice are able to form skeletal muscles if Mrf4 expres-
sion is not compromised [11]. Thus, MRF4 has roles in both
muscle determination and differentiation.
3.2 Molecular mechanisms
The bHLH transcription factors have a basic region and a
helix-loop-helix domain. These muscle-specific transcription
factors can either homodimerise, or heterodimerise with class I
bHLH transcription factors, which are quasi-ubiquitous E
proteins (such as HEB/HTF4, E2-2/ITF-2 and E12/E47). The
MRFs act on muscle-specific promoters where they can interact
with each other and with other transcription factors, such as
MEF2. In addition, they interact with chromatin-modifying
proteins, such as histone deacetylases (HDACs), histone acetyl-
transferases (HATs) and members of the SWI/SNF
ATP-dependent chromatin remodelling complex (Figure 3).
Each of the MRF members has been shown to heterodimer-
ise in vitro, and in vivo with quasi-ubiquitous E proteins that
are the alternatively spliced products of the E2A gene: E12 and
E47 [26-28]. Basically, dimers formed from bHLH proteins dif-
fer in their abilities to bind to DNA. For example, MyoD-E47
heterodimer forms efficiently and binds strongly to DNA,
whereas MyoD homodimers only poorly bind DNA. These
Table 1. The different maturation stages and
genes/proteins potentially involved at each stage
of skeletal muscle formation.
Myogenic stage Associated genetic factors
Delamination Pax3, c-Met
Migration c-Met/HGF, Lbx1, Msx1
Proliferation Pax, c-Met, Mox3, Six, (Myf5, MyoD)
Determination MRFs: Myf5, MyoD, MRF4
Differentiation MRFs (MyoD, myogenin, MRF4)
MEF2 (MEF2 A, B, C, D)
HGF: Hepatocyte growth factor; MEF: Myocyte enhancer factor;
MRF: Muscle regulatory factor; Myf: Myogenic factor; MyoD: Myogenic
determination.
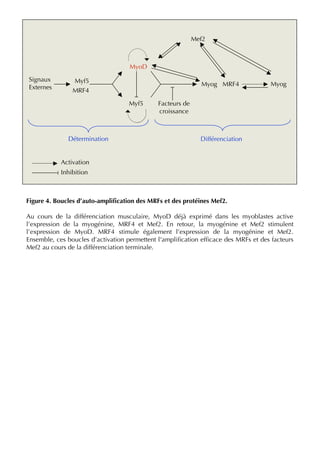
Figure 2. The MRFs functional network during muscle differentiation. MyoD, Myf5 and MRF4 are necessary for the commitment
of the somitic cells to the myogenic lineage. They are also called the myogenic determination factors. Myf5 is a positive regulator of
MyoD expression and has a direct or indirect role in myogenin gene activation. Myogenin, in turn, stimulates MyoD expression, and both
MyoD and myogenin are necessary for the late expression of MRF4 in terminal differentiation. Finally, MRF4, MyoD and myogenin
promote the expression of late stage markers such as muscle creatine kinase and myosin heavy chain.
MCK: Muscle creatine kinase; MEF: Myocyte enhancer factor; MHC: Myosin heavy chain; MRF: Muscle regulatory factor; MyoD: Myogenic determination.
External
signals
MRF4
Growth
factors
MyoD
Myf5
Determination Differentiation
MRF4Myogenin
Muscle-specific
genes
(MCK, MHC…)
Maturation](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-160-320.jpg)

![Author Proof
Yahi, Philipot, Guasconi, Fritsch & Ait-Si-Ali
Expert Opin. Ther. Targets (2006) 10(6) 5
heterodimers bind to the consensus sequence CANNTG, the
so-called E-boxes (reviewed in [29,30]). The E-boxes are located
in the regulatory regions of muscle-specific genes, such as
MCK, Desmin and Tropinin I [31,32]. E-boxes occur frequently
on the genome and not only in the regulatory region of muscle
genes. Thus, intermolecular interactions appear to be necessary
for MyoD to activate muscle gene transcription. Binding sites
for factors such as MEF2 can functionally substitute for the
second E-box, indicating that the cooperative interactions
with adjacent factors are crucial for establishing a stable and
functional transcriptional complex [33].
3.3 MyoD, the major player
MyoD is considered as the major player in triggering muscle
terminal differentiation, even if Myf5 has a redundant role
in muscle development and adult myogenesis [34]. The abil-
ity of MyoD to convert non-muscle cells into skeletal muscle
strongly indicates that it might have a central role in myo-
genesis [35,36]. These findings provided the first direct evi-
dence that a single gene can initiate a complex programme
of differentiation, acting as a ‘master switch’ as named by
S Tapscott [37]. The role of MyoD in the cell-cycle arrest and
in triggering muscle differentiation will be discussed below,
as well as the repressive complexes that inhibit MyoD activ-
ity. Indeed, the myogenic activity of the bHLHs and MEF2
factors is both positively and negatively regulated by differ-
ent chromatin remodelling enzymes [38] and ATP-dependent
chromatin remodelling complexes, which play a pivotal role
in transcriptional regulation. For example, both families of
proteins associate with HATs and HDACs, which act in an
opposing manner to control the acetylation state on
nucleosomal histones (and of the proteins themselves), in
proliferating and differentiating myoblasts.
3.4 The myocyte enhancer binding factor 2 family
The MEF-2 family is part of the MADS-box-containing pro-
teins (MCM1, agamous, deficiens, serum response factor). In
vertebrates, the four genes of the MEF-2 family are Mef2: A, B,
C and D [39], and their alternative splicing gives rise to the
MEF2 proteins. These transcription factors bind to A/T-rich
sequences found at a number of muscle-specific promoters [39].
MEF2 proteins contain the highly conserved MADS, and
the adjacent MEF2 domains, involved in DNA binding and
dimerisation [40]. MEF2 factors can directly interact with, and
help, MyoD to bind to E boxes (recognised by the bHLH fac-
tors). Indeed, in many muscle-specific genes, the MEF2 bind-
ing sites and E boxes are juxtaposed [41]. Moreover, activation
of muscle gene expression by myogenic bHLH proteins
depends on their association with members of the MEF2
family [15], as documented by experiments of loss-of-function
on Mef2 that inhibited differentiation [42,43].
The four Mef2 genes’ expression profile corresponds to that
of myogenin, suggesting that MEF2 proteins are implicated in
the final steps of muscle differentiation [44]. The Mef2c gene is
the first member of the MEF2 factors to be expressed at the
onset of differentiation of skeletal muscle lineage in vivo,
followed by the expression of the other Mef2 genes, and its
expression is maintained throughout skeletal muscle develop-
ment. It has been shown that Mef2c is a direct transcriptional
target for MEF2 and myogenic bHLH proteins [45].
4. Chromatin remodelling enzymes in
proliferating myoblasts
It is interesting to take a glance at MyoD expression through-
out the cell cycle, because MyoD is expressed in proliferating
myoblasts without triggering muscle differentiation. MyoD
levels increase during G1 phase until a restriction point where
MyoD becomes phosphorylated on serine 200 and is then
ubiquitinylated to be finally degraded. After the S phase,
MyoD levels start increasing again, but at the onset of mitosis
MyoD is phosphorylated on serines 5 and 200 by
cyclin/CDK1 complexes and is further ubiquitinylated then
degraded [46-49]. In proliferating myoblasts, MyoD actually
represses its target genes. Indeed, class I and II HDACs are
expressed in proliferating myoblasts, providing a potential
explanation for the inability of MyoD, and MEF2, to activate
their target genes prior to differentiation. There would be at
least 17 HDACs, which are organised in three classes: I, II
and III. Class I HDACs are expressed ubiquitously, whereas
class II HDACs are highly enriched in heart, skeletal muscle
and brain. Thus, in proliferating myoblasts, MyoD is associ-
ated with class I HDACs through its bHLH region [38,50,51]
(Figure 3A). This interaction represses the transcriptional activ-
ity of MyoD, in part, by deacetylating MyoD itself [51], and by
deacetylating histones on its target gene promoters [50-52]. The
dimerisation of MyoD with its co-activators E12 and E47 is
also prevented as they are heterodimerised with antimyogenic
Id protein [53]. Furthermore, the activity of MEF2 proteins is
repressed by association with class II HDACs (HDAC4 and
HDAC5), resulting in histone deacetylation on, and
transcriptional repression of, muscle genes [54].
Finally, the class III histone deacetylase Sir2, a
NAD+-dependent deacetylase, inhibits myogenesis by form-
ing a complex with the HAT p300/CBP-associated factor
(PCAF) and MyoD and, when overexpressed, retards muscle
differentiation. Conversely, cells with decreased Sir2 differ-
entiate prematurely [52].
Moreover, the recruitment of the HDAC complexes to
deacetylate histones on the muscle promoters before differen-
tiation contributes to H3K9 methylation by histone methyl-
transferases (HMTs) on these same promoters. H3K9
surrounding a MEF2 binding site in the inactive myogenin
promoter is, indeed, highly methylated in proliferating myob-
lasts, but becomes acetylated during muscle differentiation,
when the myogenin gene is activated [55]. Methylated H3K9 is
a repressive epigenetic mark, which is recognised by hetero-
chromatin protein HP1, and thus contributes to form a
heterochromatic structure that represses transcription in a
stable way (Figure 3A). The authors’ results demonstrate that](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-162-320.jpg)
![Author Proof
Chromatin modification and muscle differentiation
6 Expert Opin. Ther. Targets (2006) 10(6)
MyoD protein interacts also with HP1 in myoblasts
(unpublished observation).
Other repressive HMTs negatively regulate muscle differen-
tiation. Notably, Ezh2, which is a polycomb protein with a
HMT activity specific for histone H3K27, is recruited specifi-
cally on muscle specific genes in proliferating myoblasts [56].
Ezh2 is found in a protein complex containing HDAC1
(Figure 3A). Indeed, muscle gene promoters were found to be
hypermethylated on H3K27 and thus silenced. Triggering of
differentiation and muscle gene activation is coupled with the
loss of this repressor complex from these loci. H3K27
becomes hypomethylated and the transcriptional activators
MyoD and MEF2 are recruited, allowing the transcription of
their target genes. Downregulation of Ezh2 coincided with
activation of muscle gene expression and its hyperexpression
inhibited muscle differentiation. These findings suggest the
existence of a two-step activation mechanism, whereby
removal of Ezh2-mediated H3K27 methylation, followed by
the recruitment of positive transcriptional regulators at dis-
crete genomic loci, are required to promote muscle gene
expression and cell differentiation. The Ezh2 complex might
not regulate the expression of every muscle-specific gene.
Indeed, myogenin expression is not influenced by Ezh2 [56].
5. Chromatin remodelling enzymes in
differentiating myotubes
During the early process of skeletal muscle differentiation,
myogenic factors regulate the transition from the proliferative
to a differentiating stage. Myoblasts must irreversibly exit the
cell cycle prior to the activation of muscle-specific gene.
Studies have revealed that MyoD contributes to both of these
events (reviewed in [57]). These two functions of MyoD are
controlled by distinct mechanisms: indeed, MyoD basic region
mutants are unable to activate differentiation, but can still
induce cell-cycle arrest [58]. The key step to growth arrest
involves the downregulation of cell cycle activators such as cyc-
lins (except cyclin D3) and cyclin-dependent kinases (CDKs),
and upregulation of cell-cycle inhibitors such as retinoblastoma
protein Rb, p21, p27 and p57. Cyclin D3, together with Rb
and the CDK inhibitor protein p21, are critical cell-cycle regu-
lators for MyoD-mediated growth arrest [59,60]. These genes
share the properties of being non-muscle-specific genes already
expressed in proliferating myoblasts, but have their expression
level raised by MyoD at the onset of differentiation [60]. In dif-
ferentiating myoblasts, MyoD associates to CREB (c-AMP
responsive element binding protein) and activates the Rb pro-
moter in cooperation with the HATs p300 and PCAF [61].
Hypophosphorylated Rb promotes cell cycle arrest and the
expression of late-stage muscle-specific genes, and also prevents
apoptosis during myoblast differentiation [62,63]. On the other
hand, a functional link between MyoD, HDAC1 and Rb pro-
tein, has been provided by Puri et al. [64] (Figure 3B). Indeed,
Rb has been shown to positively regulate muscle differentiation
by competing with MyoD for binding to HDAC1. During
muscle differentiation, HDAC1 is released from MyoD and
associates with hypophosphorylated (active) Rb. Rb-HDAC1
complex appear to stimulate differentiation by repressing E2F
target genes, which are responsible of the G1/S transition [65].
Moreover, H3K9 methylation mediated by Suv39h is neces-
sary for the silencing of E2F target genes. This H3K9 methyla-
tion of the E2F target genes is a prerequisite for the induction
of terminal muscle differentiation [66]. Suv39h is indeed
required for the silencing of proliferation genes and for the
activation of the programme of muscle differentiation, and
thus it plays a major role in the control of the balance between
proliferation and differentiation.
Skeletal muscle terminal differentiation is initiated by
MyoD, which binds directly to the regulatory regions of genes
expressed during skeletal muscle differentiation and initiates
chromatin remodelling at specific promoters, such as myogenin,
MCK and the MyoD gene itself. To this end, MyoD interacts
with an adjacent protein complex containing the homeodo-
main protein Pbx1, which appears to be constitutively bound at
the muscle gene promoters [67]. Pbx1 binds constitutively to the
promoter in a SWI/SNF-independent manner, suggesting a
two-step mechanism in which MyoD initially interacts indi-
rectly with the myogenin promoter and attracts chroma-
tin-remodelling enzymes [68], which then facilitate direct
binding by MyoD and other regulatory proteins such as HATs.
The HATs/MyoD complex has two distinct HAT activities,
p300/CBP and PCAF [38,69-72], which are necessary for the
full transcriptional activity of MyoD on chromatin-associated
templates [70,73]. First, MyoD would recruit p300/CBP to the
promoter, allowing p300/CBP to acetylate histones H3
and H4. p300/CBP would then recruit PCAF (the
p300/CBP-associated factor), which acetylates three lysine resi-
dues within MyoD, thereby facilitating the transactivation
process of muscle differentiation (Figure 3B and C) [73,74].
The authors also previously showed how class II HDACs (4,
5, 6 and 7) negatively regulate muscle differentiation through
association with members of the MEF2 family [38,75]. To initi-
ate muscle differentiation, CamK, a calcium-dependant kinase
under the control of the insulin growth factor (IGF-1), triggers
the phosphorylation of two serine residues on HDAC5,
resulting in its dissociation from MEF2 Figure 3B. Then, phos-
phorylated HDAC5 associates with the 14-3-3 chaperone,
which activates a nuclear export sequence in HDAC5 and
leads to its exportation from the nucleus. The following
recruitment of p300/PCAF and other co-activators with HAT
activity to MEF2 allows muscle-specific transcription and
myotube formation [72,76].
In addition to the HATs, the SWI/SNF ATP-dependent
chromatin remodelling complex and the histone arginine
methyltransferase, co-activator-associated arginine methyltrans-
ferase 1 (CARM1), have emerged as positive key regulators of
skeletal myogenesis [55,77]. MyoD recruits the SWI/SNF
ATP-dependent chromatin-remodelling complex through an
interaction that can be regulated by the p38
MAPK Figure 3C [77,78]. At myogenic loci, the p38 kinase](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-163-320.jpg)
![Author Proof
Yahi, Philipot, Guasconi, Fritsch & Ait-Si-Ali
Expert Opin. Ther. Targets (2006) 10(6) 7
phosphorylates the SWI/SNF subunit BAF60 [78]. Moreover,
the forced activation of the p38 pathway promotes SWI/SNF
recruitment, facilitates MyoD and MEF2 binding, leads to the
recruitment of RNA polymerase II and anticipates expression of
late muscle markers at early stages of differentiation [79]. Inhibi-
tion of SWI/SNF activity, as well as inhibition of HAT activity,
prevents the ability of MyoD to initiate transcription and
chromatin remodelling at specific loci [69,77,80]. Recent results
suggest that functional SWI/SNF complexes are required for
MyoD to activate muscle-specific gene transcription, but not to
activate negative cell-cycle regulators [77].
Recent reports showed that histone arginine methylation
also plays an important role in the positive regulation of mus-
cle gene expression. CARM1, which methylates arginines, is a
positive regulator of skeletal myogenesis via direct interactions
with MEF2C Figure 3B [77]. Indeed, CARM1, which is a
member of the protein arginine–N-methyltransferase family
(PRMT) has been shown to interact with MEF2 on the
endogenous MCK promoter in a differentiation-dependent
manner. Furthermore, the inhibition of CARM1 expression
inhibits differentiation and abrogates key transcription factors
of the differentiation cascade (myogenin and MEF2). The
recruitment of CARM1 during MEF2-dependent activation
of MCK expression and its function as a MEF2 co-factor sug-
gest that histone arginine methylation (such as that of histone
H3), and non-histone targets, potentiates the process of
terminal muscle differentiation.
6. Insights from muscle regeneration studies
Studies of muscle regeneration from satellite cells have
brought more information on the different partners involved
in muscle differentiation. In response to injury, quiescent sat-
ellite cells, located between the basal lamina and the mature
differentiated muscle fibres, are activated and start proliferat-
ing to form myoblasts [81]. These myoblasts divide a limited
number of times before terminally differentiating and fusing
to form multinucleated myotubes [82,83]. The myogenic line-
age progression of satellite cells and their myoblastic progeny
is regulated by various transcription factors, including the
paired-box transcription factors Pax3 and Pax7 and the
MRFs Myf5, MyoD and myogenin [84]. Satellite cells activa-
tion from quiescence is initially characterised by upregulation
of MyoD and Myf5 expression, which precedes mitotic activ-
ity and is maintained in proliferating satellite cell-derived
myoblasts. Myogenin is also upregulated following the same
progression as in embryonic myogenesis [85]. Most of the acti-
vated cells commit to differentiation; however, a minority
downregulates MyoD, while keeping a robust expression of
Pax7, and stops DNA synthesis, thus adopting a phenotype
that resembles that of quiescent satellite cells again. This
maintenance of satellite-cell populations seems to be a uni-
versal mechanism [84]. Finally, it has been shown that MyoD
acetylation is important during muscle regeneration in vivo and
in embryonic stem cells [86]. Thus, one could speculate that a
defect in HAT enzymes responsible for MyoD acetylation
could have dramatic effects on muscle regeneration.
7. Pharmacological triggering of the
transcription regulators of skeletal muscle
differentiation
The detailed understanding of the intermolecular interactions
and the gene programmes the myogenic factors control is
essential to the rational design of therapeutics. Indeed, aber-
rant forms of myogenic factors, or their aberrant regulation,
could be associated with muscle disorders such as congenital
myasthenias, myotonic dystrophy (DM), rhabdomyosarcoma
and defects in muscle regeneration. As such, these proteins
could provide an enormous pool of target molecules where one
might intervene in muscle disorders, both with regard to
mechanisms that cause disease and regenerative mechanisms
that can ameliorate disease processes. Because transcription
factors often interact with each other as a means of both posi-
tive and negative regulation of effector genes, such interven-
tion comes with particular peril, as the results of
pharmacological manipulation are likely to be highly pleio-
tropic. Nevertheless, the manipulation of transcriptional sig-
nals holds the promise of treating muscle diseases by exploiting
the ability of muscle cells to regenerate after injury, and these
factors provide keys to unlock the incredible developmental
potential of muscle cells to repair their own damage [87].
The essential role of the MRFs in muscle development has
been proven by gene disruption experiments in mice. Mice
lacking MyoD, although overtly normal, also have defects in
muscle regeneration [88,89]. The stimulation of regeneration is
a function of MyoD that is likely to be important in
neuromuscular diseases. For example, the mdx mouse model
of Duchenne muscular dystrophy has more severe pathology
in the absence of MyoD [88].
MyoD has also been implicated in myotonic dystrophy [90].
DM is caused by two similar, non-coding, repeat expansion
mutations (DM1 and DM2). These are thought to produce
pathogenic RNA molecules that accumulate in nuclear foci.
DM1 is a CTG expansion in the 3′UTR (untranslated region
of dystrophia myotonica protein kinase [DMPK] RNA).
Introduction of a mutant DMPK 3′UTR into cultured
muscle cells disrupts muscle differentiation by lowering the
expression of MyoD [90,91], although the mechanism for this
effect is unclear.
Chromatin-remodelling factors also influence muscle differ-
entiation, hypertrophy and fibre-type determination, signalling
mechanisms that impinge on these transcriptional complexes,
and how these multicomponent regulatory cascades may be
exploited in the development of novel therapies to more effec-
tively treat myopathies in humans. Indeed, as mentioned previ-
ously, several HDACs have been shown to regulate MEF2- and
MyoD-activated transcription. HDACs 4, 5 and 7, all of which
are class II HDACs, can associate with each of the four
vertebrate MEF2 proteins and affect their function [38].](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-164-320.jpg)
![Author Proof
Chromatin modification and muscle differentiation
8 Expert Opin. Ther. Targets (2006) 10(6)
The ability of HATs and HDACs to physically interact
with MRFs and MEFs confounds the design of pharmaco-
logical agents aimed at these molecules, as perturbation of
HATs and HDACs could affect loci beyond those involved in
gene regulation in muscle. However, the fact that there are
drugs that inhibit HDAC activity might well provide novel
opportunities to stimulate myogenic gene programs. In a
recent and exciting demonstration of this idea, exposure of
mouse embryos in utero to non-teratogenic doses of HDAC
inhibitors (trichostatin A or valproic acid) increased the
number of somites in mice [92].
Chromatin remodelling was also implicated in the control
of cardiac hypertrophy (reviewed in [93]), which is often asso-
ciated with increased risk of mortality, and the molecular
mechanisms of cardiomyocyte hypertrophy could have a sig-
nificant impact on human health. Thus, it seems likely that
p300/CBP positively regulate cardiac hypertrophy. This
hypothesis should be easily testable with specific inhibitors of
p300/CBP, such as Lys-CoA and dominant-negative mutants
of the HAT, as shown by Gusterson et al. [94].
On another hand, Olson’s group results suggest key roles
for class II HDACs as negative regulators of cardiac hypertro-
phy [55], found that diverse hypertropic signals stimulate the
activity of a cardiac HDAC kinase(s) that phosphorylates the
14-3-3 binding sites in HDAC4, -5, -7 and -9, and that
signal-resistant mutants of these HDACs containing alanines
in place of the target serine residues are potent repressors of
hypertrophy. These results strongly suggest that class II
HDACs function as signal-responsive repressors of cardiac
hypertrophy and heart failure, presumably through their
capacity to inhibit MEF2 target genes.
8. Conclusion
More is now understood about how chromatin remodelling
enzymes regulate myogenesis at the molecular level. However,
much remains to be done to fully evaluate the involvement of
these enzymes in muscle disease initiation and progression. It
will be of great interest to determine their target genes in the
context of muscular homeostasis. On the basis of this infor-
mation, it should be possible to develop novel drugs and
specific treatment strategies having fewer side effects than is
currently the case.
9. Expert opinion
All of the myogenic transcription factors represent potential
targets where pharmacological intervention could have a
dramatic impact on disease processes. Agents affecting
calcium signalling, protein phosphorylation and protein
methylation could also be exploited and are another class of
target molecules for the design of therapies.
Chromatin-modifying enzymes play critical roles in the
control of muscle gene regulatory programs. A major chal-
lenge for the future is to understand how these enzymes are
coopted by signalling cascades to allow for rapid and precise
changes in gene expression in response to physiological and
pathological cues. Elucidation of these molecular mechanisms
will undoubtedly reveal novel targets that are amenable to
pharmacological manipulation in the treatment of
muscle-related diseases in humans.
In addition to traditional pharmacological approaches,
techniques such as gene therapy and RNA interference [95] are
important for manipulating this biologically important class
of molecules in muscle cells. Indeed, small interfering RNAs
(siRNA) could be inoculated into humans, in order to knock
down the expression of a harmful gene. RNA interference
gene-silencing mechanism is induced by small
double-stranded RNA and is highly sequence specific. It is an
extremely powerful tool for silencing gene expression in vitro,
and might be used as therapy in human muscle pathologies to
specifically target a pathological version of a given RNA. It
could for example suitable for use to knock down the patho-
genic DMPK RNA in myotonic dystrophy. Indeed, the use of
lentivirus-delivered short hairpin RNAs (shRNAs) seem to
downregulate the endogenous nuclear retained mutant
DMPK mRNAs in cultured cells [96].
The emerging role of small non-coding RNAs in many cell
processes, including muscle differentiation, will be of great
therapeutical interest. Some of these microRNAs, such as
miR181, are directly involved in muscle differentiation [97]. It
was, for example, recently demonstrated that a mutation in
the 3′UTR of myostatin RNA creates a binding site for two
microRNAs expressed in muscle cells causing a translation
inhibition of the myostatin gene and then contributing to
muscle hypertrophy [98]. Non-coding RNAs are linked to
chromatin remodelling and gene silencing, at least in some
organisms such as plants and fission yeast [99]. It will be of
great interest to further investigate the link between the
non-coding RNAs and chromatin remodelling in mammals.
Deciphering such a link might help in a better understanding
of chromatin remodelling in muscle physiopathology and the
design of new therapies targeting non-coding RNAs.
Acknowledgements
The authors’ laboratory is supported by the Association
Française contre les Myopathies (AFM), the Ligue
Nationale contre le Cancer (LNCC), the Association pour
la Recherche sur le Cancer (ARC), the Fondation
Bettencourt-Schueller, and the European grant
LSHG-CT-2004-502950. H Yahi and V Guasconi were
recipients of fellowships from the Ligue Nationale contre le
Cancer, and OP from Ministère de la Recherche.](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-165-320.jpg)





![Genome Biology 2007, 8:R270
Open Access!""#Robinet al./olume 2, Issue 6!, 7rticle R!#"Method
Post-translational modifications of histones H3 and H4 associated
with the histone methyltransferases Suv39h1 and G9a
:hilippe Robin=, Lauriane Fritsch=, @phélie :hilipot=, Fedor SvinarchuEF
and Slimane 7it-Si-7li=
7ddresses: =Centre National de la Recherche ScientifiLue (CNRS) FRE !PQQ, Institut 7ndré LRoff, rue GuT UoLuet, /illejuif F-PQ2"6, FranceW
Xniversité :aris-Sud, /illejuif F-PQ2"6, France. FCentre National de la Recherche ScientifiLue (CNRS) FRE Y"62, GENET[@N, bis rue de
l'Internationale, EvrT F-P6""!, FranceW Xniversité d'EvrT, EvrT F-P6""!, France.
Correspondence: Slimane 7it-Si-7li. Email: aitsiali]vjf.cnrs.fr
© 2008 Robin et al.; licensee BioMed Central Ltd.
This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which
permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
[istone modifications associated Rith [UTs^p_Uass spectrometrT analTsis of the post-transcriptional modifications of histones [Y and [Q that Rere co-purified Rith histone meth-Tltransferases SuvYPh6 and GPa shoRs that, in [eLa cells, histone methTltransferases can be phTsicallT associated Rith acetTlated histones,Rhich normallT marE transcriptionallT active chromatin.^`p_
Abstract
Specific combinations of post-translational modifications of histones alter chromatin structure,
facilitating gene transcription or silencing. Here we have investigated the 'histone code' associated
with the histone methyltransferases Suv39h1 and G9a by combining double immunopurification and
mass spectrometry. Our results confirm the previously reported histone modifications associated
with Suv39h1 and G9a. Moreover, this method allowed us to demonstrate for the first time an
association of acetylated histones with the repressor proteins Suv39h1 and G9a.
Background
The amino-terminal tails of nucleosomal histones protrude
from the aN7 and are subject to covalent modifications.
These modifications include lTsine acetTlation, lTsine and
arginine methTlation, serine and threonine phosphorTlation,
7a:-ribosTlation, and ubiLuitination c6d. [istone lTsine
methTlation can have different effects depending on the resi-
due that is modified: methTlation of histone [Y at LTsQ
([YeQ) is associated Rith gene activation, Rhereas methTla-
tion of [YeP, [Ye!#, and [Qe!" generallT correlates Rith
transcriptional repression c!-Qd. The roles of [YeYf and
[Ye#P methTlation remain elusiveW indeed, these modifica-
tions are associated Rith both transcriptional activation and
repression cg,fd.
LTsine residues can be mono-, di-, or trimethTlated, inducing
different biological responses cY,#,2d. Thus, for example,
highlT condensed heterochromatic regions shoR a high
degree of trimethTlated [YeP ([YePmeY), Rhereas euchro-
matic regions are preferentiallT enriched in mono- and
dimethTlated [YeP c!,Yd. [istone lTsine methTlation is
mediated bT histone methTltransferases ([UTs), manT of
Rhich contain a conserved SET cSu(var)Y-P, Enhancer-of-
zeste, Trithoraxd domain, such as SuvYPh6 (Suppressor of
variegation YPh6) and GPa c6,!,Pd. SuvYPh6 belongs to a fam-
ilT of peri-centromeric proteins and is responsible for [YeP
trimethTlation c6"-6Yd. GPa (Eu[UTase-!) is the major
methTlase responsible for mono- and dimethTlation of [YeP
in euchromatic regions c6Q,6gd, but it maT also be present in
heterochomatic regions c6fd.
Covalent modifications of histones can regulate gene expres-
sion directlT or through recruitment of non-histone effector
proteins c!,6#d. These effector proteins bind modified chro-
matin using a varietT of chromatin-binding domains. For
example, bromodomains recognize acetTlated lTsines,
Rhereas chromo, UBT, Tudor, kQ" domains and :[a fin-
gers, recognize methTlated lTsines c6#,62d. Repressive
Published: 20 December 2007
Biology 2007, 8:R270 (doi:10.1186/gb-2007-8-12-r270)
Received: 2 October 2007
Revised: 6 December 2007
Accepted: 20 December 2007
The electronic version of this article is the complete one and can be
found online at http://genomebiology.com/2007/8/12/R270](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-171-320.jpg)

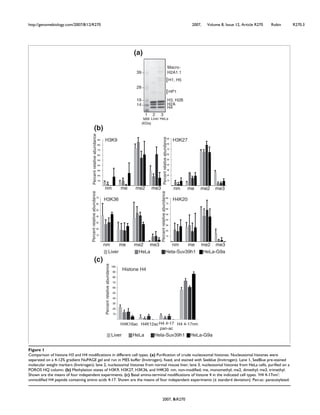

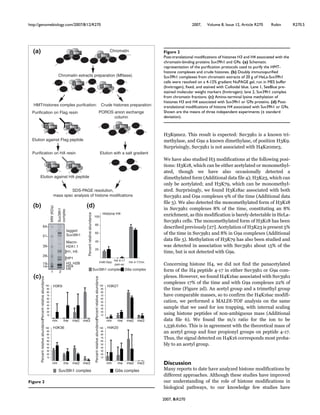








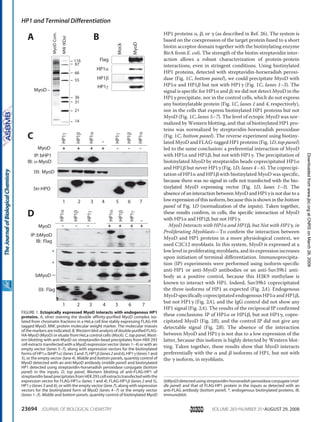

![(Fig. 6A). Overexpression of any of the HP1 isoforms, ␣, , or
␥, resulted generally in the inhibition of differentiation com-
pared with the control cell line. HP1-overexpressing cells did
not express the differentiation markers myogenin and MCK
(Fig. 6B) and did not fuse into myotubes (data not shown).
The faint expression of MCK and myogenin in HP1␣-ex-
pressing cells cultured in differentiation medium was due to
the fact that this cell line was not pure for the expression of
tagged HP1␣, ϳ90%; during cul-
ture, some cells lost tagged HP1␣
expression. We presume that
these cells differentiated normally
and expressed myogenin and
MCK.
Overexpression of HP1 proteins
could have broad effects (31). Thus,
differentiation inhibition we have
seen in cells overexpressing HP1
proteins could be a result of these
broad effects. To check the specific-
ity of the effects seen when we over-
express HP1 isoforms (Fig. 6, A and
B), we performed ChIP experiments
using anti-FLAG resin to test
whether the tagged HP1 isoforms
are recruited to MyoD target genes.
Our results show that the exoge-
nous HP1 isoforms are indeed phys-
ically recruited into MyoD target
genes (Fig. 6C), but unlike the
endogenous HP1 (Fig. 5), these
exogenous isoforms remain on
MyoD target promoters even in
myoblasts cultured in differentia-
tion conditions (Fig. 6C). This result
suggests that the differentiation
defect seen in HP1-overexpressing
myoblasts could be due, at least in
part, to a lack of de-repression of
MyoD target genes. Taken together,
these results suggest that overex-
pression of HP1 proteins impairs
MyoD target genes expression and
thus muscle terminal differentia-
tion, regardless of the HP1 isoform.
DISCUSSION
Repression of MyoD target genes
in proliferating myoblasts involves
H3K9 methylation (20, 22), which is
normally recognized by HP1 pro-
teins (4, 5). This suggests the forma-
tion of local facultative heterochro-
matin on MyoD target promoters,
insuring their stable repression
until the cells undergo terminal dif-
ferentiation. We tested the hypoth-
esis that HP1 proteins are involved
in the repression of MyoD target genes and explored their role
in muscle terminal differentiation in general.
Physical and Functional MyoD/HP1 Interaction—Immuno-
precipitation assays showed that MyoD interacts with HP1␣
and HP1, but not with HP1␥ in myoblasts. In vitro assays dem-
onstrated that MyoD interacts directly with HP1␣ and HP1
via their chromoshadow domains and the MyoD transactivat-
ing domain. A reporter gene assay indicated that this interac-
A
IB:MyoD
IB:GST
GST-HP1α
GST-HP1β
GST-HP1γ
GST
1 32 4
WT
119-189
1-119
67-119
1-67
C
IB:MyoD
IB:GST
CD CSD
1 67 119 189
HP1α
1 32 4 5
GST-HP1α:
MyoDWT
MyoDWT
Cteraa173-318
Nteraa1-114
∆Cteraa1-241
∆Nteraa83-318
GST-HP1βGST
Emptyvector
α-Flag
α-GST
GST-HP1β
GST
α-HA
D
1 31899 167
CterNter bHLH
TAD
Dimerization
DNA binding TAD
MyoD
GST
GST-HP1α
GST-HP1β
GST-HP1γ
GST
GST-HP1α
GST-HP1γ
input
input
GST-HP1α
GST-HP1γ
GST-HP1β
GST-HP1α
GST-HP1γ
+ --- --+ + + +++
1 2 3 4 5 6 7 8 9 10 11 12 13 14
MyoDMyogenin
MyoDMyogenin
B
MyoDWT
Cteraa173-318
Nteraa1-114
∆Cteraa1-241
∆Nteraa83-318
Emptyvector
E
*
*
*
* *
35,1
47,4
24,9
24,9
35,1
47,4
35,1
47,4
60,4
24,9
MW
KDa
MW KDa
MW KDa
y
MW KDa
35,1
*
*
*
*
*
47,4
35,1
24,9
47,4
60,4
24,9
35,1
47,4
24,9
47,4
60,4
FIGURE 3. HP1␣ and HP1, but not HP1␥, interact directly with MyoD in vitro via their chro-
moshadow domain and MyoD transactivating domain. A, HP1␣ and HP1 interact directly with MyoD
in a GST pulldown assay. Equivalent amounts of bacterially produced untagged MyoD were incubated
with GST beads (lane 4) or GST-HP1 beads (lanes 1–3). MyoD was detected by Western blotting using an
anti-MyoD antibody (upper panel), and GST fusions using an anti-GST antibody (lower panel). IB, immuno-
blot. B, unlike MyoD, myogenin does not interact with HP1 proteins. Myogenin and MyoD were in vitro
translated in the presence of radioactive [35
S] methionine (inputs on lanes 1 and 14, respectively). MyoD or
myogenin were incubated with equivalent amounts of either GST or GST-HP1 proteins, as indicated. GST
pulldown was then conducted as described under “Experimental Procedures,” except that at the end of
the experiment, the radiolabeled proteins were detected by autoradiography. Lanes 1–8, GST pulldown
experiment with [35
S]myogenin; lanes 9–14, with [35
S]MyoD. C, the chromoshadow domain of HP1␣ is
required for interaction with MyoD. Equivalent amounts of bacterially produced MyoD were incubated
with GST-HP1␣ deletion mutants on beads (lanes 2–5) or GST-HP1␣ wild type (lane 1). MyoD was detected
by Western blotting using an anti-MyoD antibody (upper panel) and GST fusions using an anti-GST anti-
body (lower panel). D, HP1 interacts with MyoD wild type and with MyoD deletion mutants containing
the C-terminal domain. Top panel, schematic diagram of MyoD functional domains . TAD, transactivation
domain. Lower panels, wild type MyoD was detected using anti-FLAG antibody, and its deletion mutants
were detected using anti-HA antibody (upper panels). GST and GST-HP1 were detected using anti-GST
antibody (lower panel). E, Western blotting using anti-FLAG and anti-HA to verify the expression level of
MyoD and its deletion mutants in HeLa cells used in D*
, specific bands.
HP1 and Terminal Differentiation
23696 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283•NUMBER 35•AUGUST 29, 2008
atCNRSonMarch26,2009www.jbc.orgDownloadedfrom](https://image.slidesharecdn.com/1cadc5b6-0df2-4ecd-a684-c949097bc1c4-150604075202-lva1-app6892/85/manuscrit-these-Ophelie-Philipot-186-320.jpg)