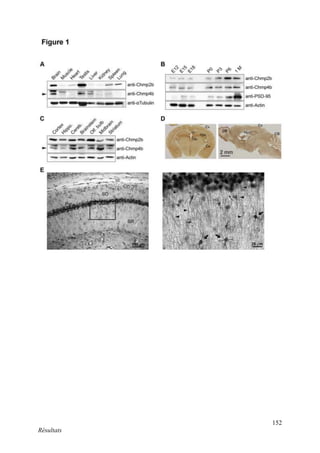
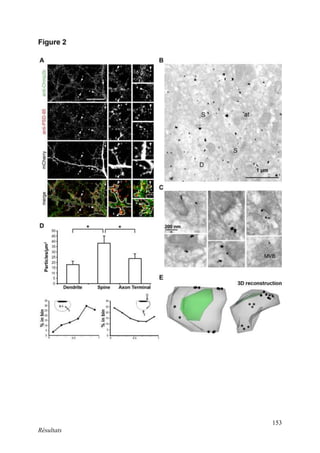
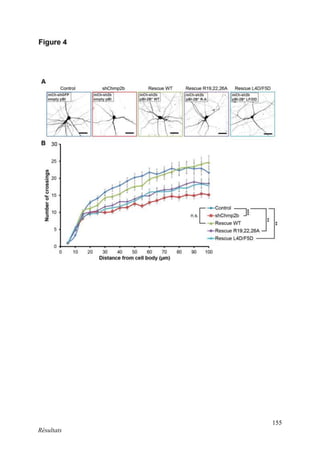
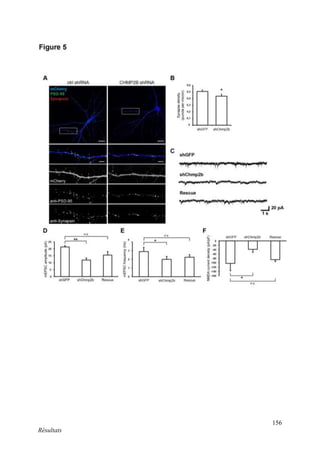
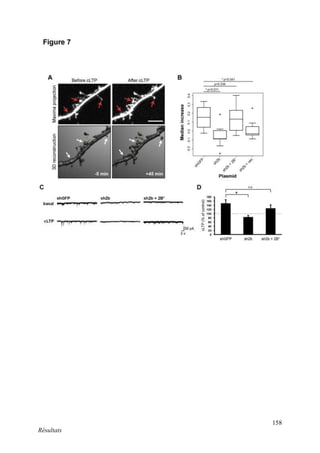
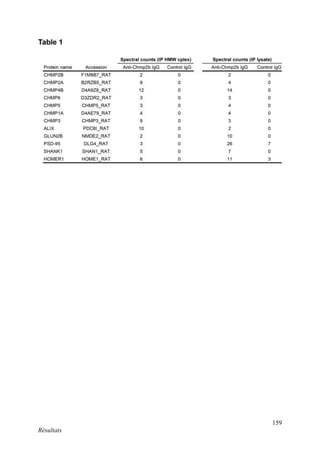
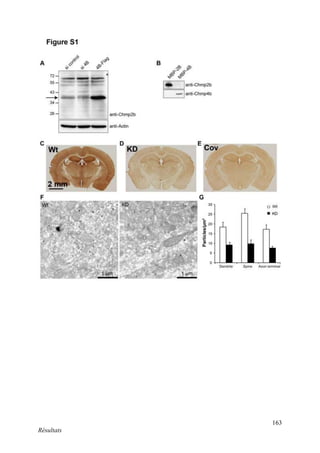
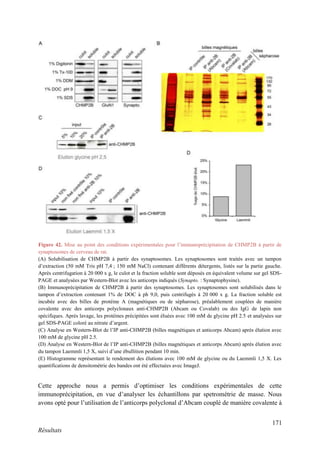
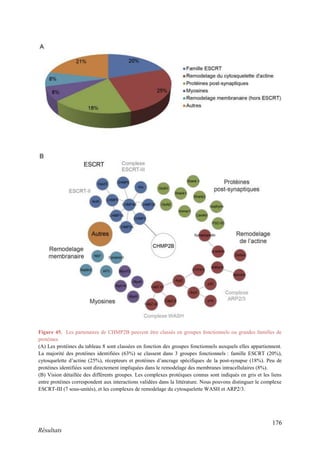
Cette thèse présentée par Romain Chassefeyre se concentre sur le rôle de la protéine CHMP2B et du complexe ESCRT-III dans le remodelage des membranes cellulaires, notamment dans le développement des épines dendritiques et leur implication dans les maladies neurodégénératives. Soutenue publiquement le 16 décembre 2013, la recherche explore les mécanismes de déformation membranaire et leur relation avec des pathologies telles que la démence frontotemporale. Le document inclut également des remerciements aux membres du jury et à l'équipe de recherche, ainsi qu'une description détaillée des méthodes et résultats obtenus dans le cadre de l'étude.

























![18
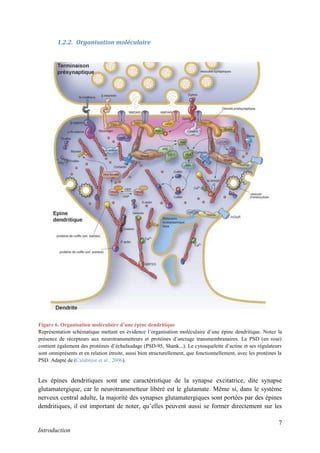
Introduction
Nous verrons que le maintien à long terme de la potentialisation passe par des modifications
profondes du fonctionnement et de la morphologie des épines dendritiques.
Le Ca2+
libéré par l’ouverture du récepteur NMDA est considéré comme le second messager
responsable de l’induction de la LTP. De nombreuses études ont montré l’importance de
l’augmentation de la concentration postsynaptique en Ca2+
([Ca2+
]ED) pour le déclenchement
de la LTP. L’injection intracellulaire d’EGTA, un chélateur des ions divalents, empêche la
potentialisation (Lynch et al., 1983), alors qu’à l’inverse, l’augmentation de la [Ca2+
]ED à
tendance à potentialiser les synapses. Comme nous l’avons vu, la morphologie spécifique des
épines dendritiques limite la diffusion des molécules vers la dendrite et permet de maintenir
une [Ca2+
]ED élevée au niveau de la tête de l’épine (Noguchi et al., 2005). Le Ca2+
déclenche
la LTP en activant des cascades de signalisation au niveau postsynaptique qui vont aboutir à
des modifications post-traductionnelles de protéines. Il active notamment la CamKII
(Ca2+
/Calmodulin dependent kinase II), protéine la plus abondante dans les épines
dendritiques, ainsi que la PKC (Protein Kinase C) (Figure 13). Ces deux kinases sont
fondamentales pour l’induction de la LTP (Malinow et al., 1988; Silva et al., 1992). Elles
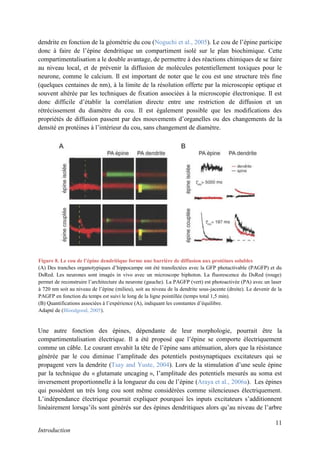
Figure 13. Le Ca2+
est l’élément déclencheur de la LTP
L’augmentation de la concentration en Ca2+
, dans la tête de l’épine, via l’ouverture des NMDA-R, va déclencher
des cascades de signalisation (CaMKII, PKC...) qui aboutissent à l’augmentation du nombre et de la
conductance des AMPA-R. Adapté de (Purves, 2004), page 589.](https://image.slidesharecdn.com/ae6d2edd-e0a5-47e6-b9a5-09a4930d6a93-170123021735/85/R-Chassefeyre-Dissertation-in-French-28-320.jpg)






























































































































































































![[0] maitre exclave](https://cdn.slidesharecdn.com/ss_thumbnails/0maitreexclave-121125111144-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)












