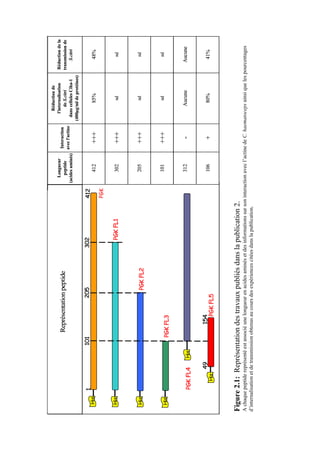
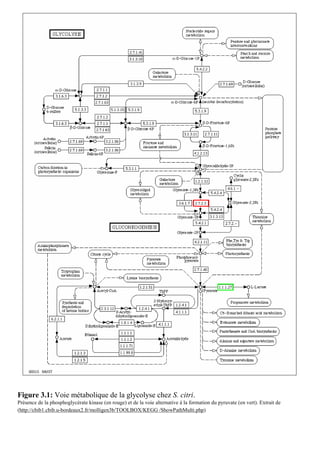
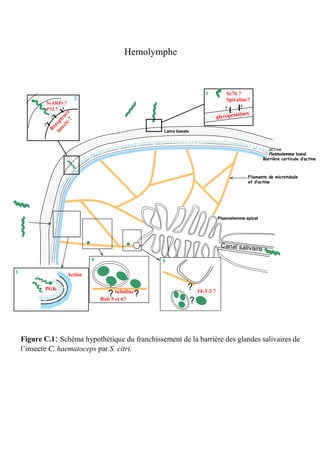
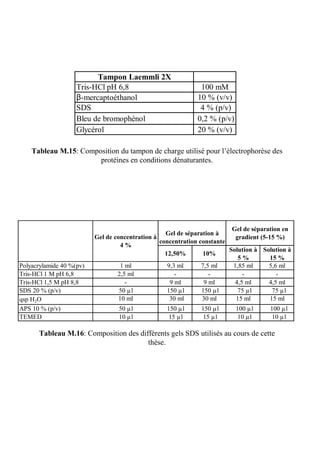
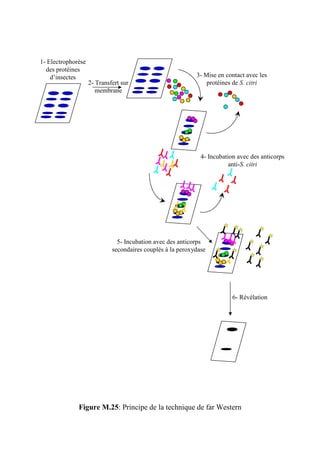
La thèse de Fabien Labroussaa, soutenue le 20 décembre 2010 à l'université Victor Ségalen Bordeaux 2, explore les interactions entre Spiroplasma citri et son vecteur insecte Circulifer haematoceps. Elle met en évidence le rôle de la phosphoglycérate kinase de S. citri, une protéine liant l'actine, dans la transmission du spiroplasme par la cicadelle. Ce travail a été réalisé au sein de l'unité mixte de recherche 1090 consacrée à la génomique et à la diversité du pouvoir pathogène.





































































































![1880 LABROUSSAA ET AL. APPL. ENVIRON. MICROBIOL.
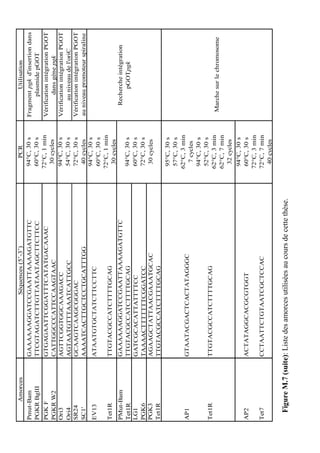
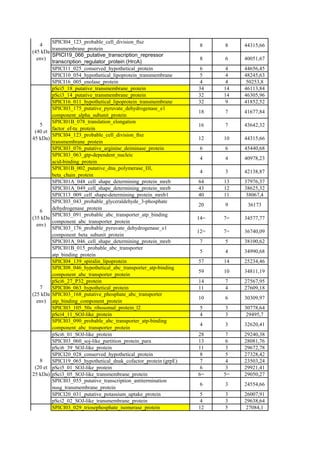
These proteins are encoded by plasmids pSci1 to -6 (46), which TABLE 1. Primers used for site-directed mutagenesis
are present only in transmissible strains, and ScARPs share of S. citri PGKa
strong similarities with the adhesion-related protein SARP1 of Primer Sequence (5Ј33Ј)b Positionc
S. citri strain BR3, in which the presence has been correlated to
PGK-F GTGAGAATTCGGATTTCATATG Ϫ19 to ϩ9
the ability for the spiroplasma to adhere to insect cells in vitro ACAAAC
(9, 55). The specific interactions of S. citri with eukaryotic cells PGK-W1F CTTTAGGGAAATATTGGGCAAG 449–471
remain to be elucidated, but a combination of the effects of PGK-W1R CTTGCCCAATATTTCCCTAAAG 449–471
several proteins or a complex would be necessary to explain the PGK-W2F GTTACTTGGAATGGGCCAATG 991–1011
invasion of a variety of host cell types by S. citri (33). PGK-W2R CATTGGCCCATTCCAAGTAAC 991–1011
PGK-R TTATGAAGCTTTTATTTACTTTG 1222–1250*
Nevertheless, in the last sequence of events involved in insect AACAGC
vector transmission, the first contact and recognition for the effi-
a
All regions were amplified using the respective mutagenic forward and re-
cient penetration of the salivary gland cells represents an essential verse primers, as required. Overlapping fragments with changes were then an-
step. In the present study, confocal images of infected salivary nealed and amplified using PGK-F and PGK-R to generate full-length products.
b
glands show the localization of S. citri cells along the actin fila- Introduced EcoRI, HindIII, and NdeI sites are underlined, and changed
nucleotides are shown in bold.
ments. We report the results of the first attempt to decipher the c
A negative value indicates an position upstream from the adenine nucleotide
role of the spiroplasma’s phosphoglycerate kinase (PGK) in the of the starting ATG codon. *, 11 nucleotides are downstream of the stop codon
internalization of S. citri in its insect vector’s cells. located at position 1239.
MATERIALS AND METHODS
immersed in fixative (4% paraformaldehyde in PBS plus 0.2% Triton X-100)
Spiroplasma strain, leafhoppers, and cell line culture. S. citri GII3, originally overnight at 4°C. Then, fixed salivary glands were rinsed three times with PBS
Downloaded from aem.asm.org at INRA on April 6, 2010
isolated from its leafhopper vector C. haematoceps captured in Morocco (52), with 0.2% Triton X-100 and permeabilized with PBS containing 1% Triton
was cultivated in SP4 medium (50) at 32°C. X-100 (PBS-T) overnight at 4°C followed by an incubation in blocking buffer
Healthy C. haematoceps leafhoppers were reared in an insect-proof cage on (PBS-T plus 1% bovine serum albumin [BSA]) for 1 h at RT. S. citri polyclonal
stock plants (Mattiola incana) at 30°C. Microinjection of S. citri GII3 into C. antibodies diluted 1:500 in PBS containing 1% BSA were added for 2 h. They
haematoceps has been described previously (21). Injected leafhoppers were were washed in PBS and treated with Alexa 488-conjugated goat anti-rabbit IgG
maintained for 2 weeks on stock plants before salivary glands were removed. (Invitrogen) at a 1:200 dilution, and simultaneously F-actin was stained by Alexa
The nonphagocyte cell line Ciha-1 (Circulifer haematoceps 1) (18) was cultured 568-phalloidin (Invitrogen) at a 1:40 dilution in PBS containing 1% BSA for 1 h
at 32°C in Schneider’s Drosophila medium (Invitrogen) supplemented with 1% at RT. After three washings in PBS, they were then mounted with ProLong Gold
histidine buffer (0.057 M histidine monohydrate, pH 6.2), 10% Grace’s insect cell antifade reagent (Invitrogen). The specificity of immunostaining was evaluated
culture medium, 0.5% G-5 supplement, and 10% (vol/vol) heat-inactivated fetal by omitting the antibodies against S. citri proteins. Immunofluorescence samples
bovine serum. were imaged using a TCS SP2 upright Leica confocal laser scanning micro-
Far Western blotting experiments. Leafhopper salivary glands were dissected scope, with a 40ϫ water immersion or 63ϫ oil immersion objective lens at
in phosphate-buffered saline (PBS; 2 mM KH2PO4, 8 mM Na2HPO4, 0.14 M 1,024 by 1,024 pixel resolution. The images were coded green (Alexa 488) and
NaCl, 2 mM KCl [pH 7.4]) containing 1 mM phenylmethanesulfonylfluoride red (Alexa 568).
(PMSF). Glands were stored frozen in buffer at Ϫ20°C until use. Glands were Partial purification of S. citri proteins involved in an interaction with actin.
transferred to a potter Elvehjem grinder containing the same buffer and homog- Polyclonal antibodies against chicken actin (Sigma) were linked to protein
enized. Then the mixture was centrifuged for 1 min at 10,000 ϫ g. The protein A–Sepharose CL-4B according to the manufacturer’s manual (GE Healthcare).
concentration was determined by the Bradford procedure. Proteins were not ¨
All steps were conducted on an AKTA purifier liquid chromatography system
further purified, to avoid inadvertently removing proteins of interest. Aliquots of (GE Healthcare). Noninfected leafhopper proteins were prepared as described
supernatant (20 g) were fractionated by electrophoresis in a 12.5% SDS-PAGE above for salivary gland proteins. To trap leafhopper actin with antiactin anti-
gel before transfer onto a nitrocellulose membrane according to the methods of bodies, 1 mg of leafhopper proteins was loaded on the column and proteins
Killiny et al. (32). After transfer, all steps were conducted under low agitation. bound nonspecifically to the actin column were eliminated with a 10-ml step
Membranes were blocked in 10 ml of PBS with 6% nonfat dry milk and incubated using 2 M NaCl. Then, 500 g of S. citri proteins, prepared as described in the
with 2 ml of S. citri proteins (20 g/ml) in PBS overnight at 4°C. S. citri proteins previous section for far Western experiments, was loaded on the actin column. S.
used as an overlay were prepared according to the methods described by Killiny citri proteins bound to actin were eluted with a 0 to 2 M NaCl gradient (total
et al. (32) with a few modifications. Disruption of the cells was performed by volume, 20 ml). All the eluted proteins were subjected to SDS-PAGE, followed
sonication (Vibracell sonicator; Sonics & Materials, Inc., Danbury, CT; rate of by gel staining or far Western analysis. Nonspecific binding of S. citri proteins on
40% pulses/s, 50 W, 4°C; 1 min of sonication and 1 min on ice alternatively, three protein A-Sepharose and/or on antiactin antibodies was highlighted by a control
times). After incubation, blots were washed with 50 ml of PBS buffer containing experiment carried out without C. haematoceps proteins loaded on the column.
0.1% Tween 20 (PBS-Tween) and incubated in 10 ml of PBS containing 1% S. citri proteins eluted from such columns were also subjected to SDS-PAGE and
nonfat dry milk (antibody buffer) with purified polyclonal IgGs against total S. far Western blotting. Far Western experiments were carried out according to the
citri proteins at a final concentration of 5 g/ml for 1 h at room temperature conditions described above. Blots of eluted proteins were incubated with a
(RT). After three washings with PBS-Tween, blots were incubated in 10 ml of mixture of C. haematoceps proteins, and the binding activities were revealed with
antibody buffer with peroxidase-conjugated goat anti-rabbit IgGs (Sigma Al- polyclonal antibodies against actin. Gels intended for mass spectrometry analysis
drich) at a 1:50,000 dilution at RT for 1 h. Blots were then washed in PBS-Tween were stained with colloidal blue (38).
three times, followed by incubation with the substrate solution (Super Signal Protein identification by LC-MS/MS. Proteins of interest were excised from
West Pico chemiluminescent substrate) according to the manufacturer’s instruc- stained gel and digested with trypsin as previously described (31). The resulting
tions (Pierce, Rockford, IL). Then blots were exposed on X-ray film. digestion was used for peptide mass fingerprinting by liquid chromatography-
Two micrograms of S. citri purified recombinant PGK was used in a far tandem mass spectrometry (LC-MS/MS) as routinely performed on the proteo-
Western assay carried out to confirm interaction with leafhopper actin protein. mics platform of the University of Bordeaux 2, France (22).
The blot of His6-tagged PGK was overlaid with 500 g of total insect proteins. Expression and purification of S. citri phosphoglycerate kinase recombinant
The experiment was conducted in a manner similar to that described above for protein. S. citri uses the UGA opal codon to incorporate tryptophan rather than
the S. citri protein overlay assay except that anti-His monoclonal antibodies a stop codon as in the universal genetic code (16). For PGK expression in
(MAb; Sigma) instead of S. citri polyclonal antibodies were used. Escherichia coli, the two opal codons contained in the gene were changed by
Immunofluorescence analysis of salivary glands. Salivary glands from infected site-directed mutagenesis by using overlap extension PCR (23). Primers used in
or noninfected C. haematoceps leafhoppers were dissected and incubated in 500 the PCR are presented Table 1. The amplified product of 1,239 bp corresponding
l of PBS containing 0.2% Triton X-100 at RT. All subsequent steps were to the pgk gene, in which the two opal codons were replaced by TGG codons, was
performed in the same volume. Salivary glands were washed twice in PBS and inserted in a plasmid (pBS) and the construct was used to transform E. coli](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-113-320.jpg)

![VOL. 76, 2010 S. CITRI PGK AND LEAFHOPPER ACTIN INTERACTION 1881
DH10B. The gene of interest was then transferred into a plasmid pET28a(ϩ) were fixed for 15 min in 500 l of 4% formaldehyde at room temperature,
vector (Novagen), and 1 g of the recombinant plasmid (pET28 FL) was used to washed three times with 500 l of PBS, and permeabilized by incubation for 15
transform E. coli DH10B. The desired sequence of the resulting plasmid (pET28 min with 500 l of 0.1% Triton X-100 in PBS–1% BSA followed by another three
FL) was verified by sequencing the insert. Two micrograms of the pET28 FL times wash with 500 l of PBS. Anti-His MAb at a 1:1,000 dilution was then
plasmid bearing the two desired mutations was used to transform E. coli added, followed by a secondary Alexa 514-conjugated rabbit anti-mouse antibody
BL21(DE3). Transformants were selected on Luria-Bertani (LB) solid medium (1:200). Actin was stained with Alexa 568-phalloidin (1:40) and nuclei with 0.2
containing kanamycin (50 g/ml) at 37°C. g/ml of DAPI. The coverslips were rinsed once in water and were mounted onto
To check the expression of the PGK fused to the N-terminal hexahistidine slides as described above for salivary gland observations.
sequence under the control of an isopropyl--D-thiogalactopyranoside (IPTG)- The images were coded with blue (DAPI), green (Alexa 488), and red
inducible promoter, one transformant was cultivated in 500 ml of LB medium (Alexa 568).
with kanamycin (30 g/ml) until late log phase (optical density at 600 nm, 0.6).
The recombinant His6-tagged PGK protein was produced at 28°C in culture by
adding 0.1 mM IPTG for 3 h. Bacterial cells were centrifuged at 7,000 ϫ g for 10 RESULTS
min at 4°C, and then the pellet was resuspended in 10 ml of lysis buffer (50 mM
Tris-HCl [pH 8.0], 0.3 M NaCl, 25 mM MgSO4, 2.5 mM MnCl2, 1 mM PMSF,
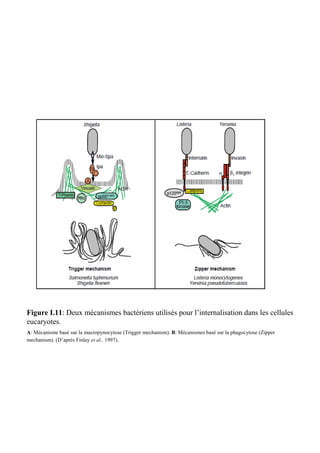
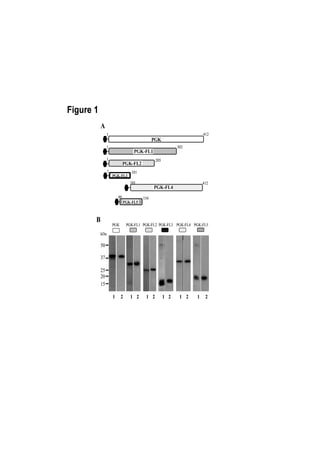
In vitro protein interaction and in vivo colocalization of S.
and 100 unit/ml of DNase I). After 10 min of incubation at RT, lysozyme was citri with host cytoskeleton. Salivary gland proteins from
added at 0.2 mg/ml for 10 min. The mixture was lysed by sonication (rate, 50% healthy leafhoppers separated by SDS-PAGE in one dimen-
pulses/s; 50 W; 4°C; 1 min of sonication and 1 min on ice alternatively) until a sion were stained with colloidal blue (Fig. 1A, lane 1) or blot-
clear lysate was obtained. The insoluble proteins were removed by subsequent ted onto nitrocellulose and probed with S. citri proteins. Bind-
centrifugation at 13,000 ϫ g at 4°C for 10 min. The recombinant protein was
purified by affinity chromatography using Ni2ϩ-nitrilotriacetic acid (Ni-NTA)
ing levels were detected with polyclonal IgGs against total S.
prepacked columns (Qiagen) followed by cation exchange chromatography citri proteins. Spiroplasma proteins were found to bind to a set
¨
(Mono Q columns; GE Healthcare) on the AKTA purifier liquid chromatogra- of insect salivary gland proteins having apparent molecular
phy system (GE Healthcare) according to the manufacturer’s instructions. masses of 42, 35, 30, 27, and 25 kDa (Fig. 1A, lane 2). In the
The purified recombinant His6-tagged PGK protein was tested with anti-His
control experiment, in which S. citri proteins were omitted
Downloaded from aem.asm.org at INRA on April 6, 2010
MAb and antiactin polyclonal antibodies in a Western blot experiment con-
ducted as described previously (19). from the overlay assay, no leafhopper protein was recognized
Effect of PGK on S. citri attachment and invasion. (i) Attachment of S. citri to by polyclonal IgGs (Fig. 1A, lane 3). The protein band with a
Ciha-1 cells. Monolayers of Ciha-1 cells grown in 24-well plates (approximately mobility of ϳ42 kDa was excised from the stained gel, washed,
2 ϫ 105 cells per well) were incubated with 900 l of various concentrations of and digested with trypsin prior to LC-MS/MS analysis. The MS
PGK from Saccharomyces cerevisiae (Sigma) or tagged PGK from S. citri in
culture medium. Cells without PGK treatment were the positive controls, and
spectra matched, in the NCBI nonredundant protein database,
uninfected cells served as negative controls. As additional controls, the effects of those of actin from the fruit fly (Drosophila melanogaster),
BSA (Sigma) and those of His6-tagged eukaryotic initiation factor 4E (eIF4E) brine shrimp (Artemia sp.), tapeworm (Diphyllobothrium den-
protein on S. citri adhesion were also assayed. Each assay condition was evalu- driticum), and silk moth (Bombyx mori). The finding that actin
ated in triplicate. After incubation with proteins for 2 h at 32°C, the cells were
was involved in an in vitro interaction with S. citri proteins
infected with a 100-l S. citri culture at a multiplicity of infection (MOI) of 15 to
30 for 4 h at 4°C. Normally, this temperature allows spiroplasma attachment to suggested that in vivo an interaction between S. citri and the
the cell surface but inhibits eukaryotic cell processes required for internalization. insect actin cytoskeleton may be involved in spiroplasma inva-
Following the incubation, the cells were then washed three times with 500 l of sion of cells.
Schneider’s Drosophila medium to remove any spiroplasmas that had not at- Thus, the far Western results prompted us to explore by
tached to the monolayer. After trypsinization for 10 min at 32°C with TrypLE
(Invitrogen), dilutions of the cells associated with adherent spiroplasmas were
confocal laser scanning microscopy the location of S. citri in
directly plated on solid SP4 medium. After incubation at 32°C for 1 week, the salivary glands. In those infected with S. citri (Fig. 1B), spiro-
number of spiroplasma colonies on each plate was counted to estimate the plasmas (green fluorescence) are preferentially present along
number of Ciha-1 cells that had adherent spiroplasmas (4). In addition, treat- the actin filaments (red color). Salivary glands from nonin-
ment of spiroplasmas with trypsin alone in the absence of Ciha-1 cells had no
fected leafhoppers did not show spots of green fluorescence
effect on spiroplasma growth (18). The relative percentage of adhesion was
calculated as follows: [(number of CFU for cells with protein treatment)/(num-
specific to spiroplasmas (Fig. 1B).
ber of CFU for untreated control cells)] ϫ 100%. For statistical analysis, Stu- Identification of one S. citri protein interacting in vitro with
dent’s t test was used when appropriate. actin. To determine the S. citri proteins interacting with actin,
(ii) Spiroplasmal entry analysis. The effect of PGK treatment on the inter- spiroplasma proteins were loaded on a column of leafhopper
nalization of S. citri into Ciha-1 cells was determined as previously described
actin that was trapped by antiactin antibodies linked to protein
using the gentamicin protection assay (4, 28). Until S. citri infection, all the steps
were the same as those described above for attachment. Infection by 100 l of S. A-Sepharose. The presence of actin in the mixture of leafhopper
citri culture at an MOI of 15 to 30 was carried out at 32°C for 18 h. The cells were proteins used to prepare the column (Fig. 2A, lane 1) was con-
thereafter washed three times with 500 l of Schneider’s Drosophila medium firmed by Western blotting using antiactin antibodies (Fig. 2A,
under low agitation to remove unbound bacteria. In order to eliminate bacteria lane 2).
that had attached but not internalized, cells were incubated with 1 ml of fresh
culture medium containing 400 g/ml of gentamicin (10 times the MIC) for 3 h
As shown in Fig. 2A, lane 3, S. citri proteins eluted from the
at 32°C. Gentamicin was eliminated by three washes with 500 l of Schneider’s antiactin–protein A-Sepharose column were those captured on
Drosophila medium followed by three additional washes with the same volume Sepharose and/or on actin antibodies (control experiment).
of PBS (1.54 mM KH2PO4, 155.17 mM NaCl, 2.71 mM Na2HPO4 ⅐ 7H2O; pH Figure 2A, lane 4, shows the profile of S. citri proteins eluted
7.2). After washes, a 100-l aliquot of the last wash was plated on SP4 medium
from the actin column. Comparison of the two protein patterns
to ensure that all extracellular bacteria had been killed (data not shown). Ciha-1
cells were trypsinized, plated on SP4 medium, and incubated at 32°C for 1 week. revealed one protein at 44 kDa present among the proteins
The number of colonies on each plate was counted to determine the number of bound to actin but absent in the control profile. To determine
cells in which S. citri was internalized. whether this protein displayed an affinity for actin, a far West-
Binding of His6-tagged PGK to Ciha-1 cells. To determine whether the contact ern assay was performed using as overlay a mixture of whole
with Ciha-1 cells of His6-tagged PGK, BSA, and His6-tagged eiF4E has an effect
on actin cytoskeleton, cells were grown on coverslips in 24-well plates and
leafhopper C. haematoceps proteins containing actin. A signif-
prepared by the same process described above until infection with S. citri. After icant binding activity located at approximately 44 kDa was
the three washes in 500 l of Schneider’s Drosophila medium, cells on coverslips revealed by rabbit antiactin IgGs followed by goat anti-rabbit](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-115-320.jpg)







































![Chapitre 2
phenylmethanesulphonylfluoride (PMSF) attachment to and internalization into Ciha-
and homogenized. Then the mixture was 1 cells were estimated by counting
centrifuged twice for 1 min at 500 × g. colonies on solid SP4 medium as described
Protein concentration in the supernatant previously (Labroussaa et al., 2010).
was determined by the Bradford procedure Gentamicin protection assay was used for
and aliquots of 500 µg of proteins were the invasion assay. Briefly, cells were
used as overlay for incubation with the incubated with 400 µg/ml of gentamycin
blots of His6-tagged proteins. Bindings during 3h at 32°C in order to kill
were revealed with rabbit polyclonal extracellular bacteria.
antibodies against actin (1:600 dilution,
Sigma) followed by goat anti-rabbit S. citri experimental transmission assays
labelled with peroxidase (1:50,000 Female leafhoppers were micro-injected
dilution, Sigma). All the steps were with 0.1 µl of a spiroplasma culture (108
conducted in the same conditions that spiroplasmas /ml) and caged on healthy
those previously published (Labroussaa et stock (Matthiola incana) plants at 30±2°C
al., 2010). as described previously (Foissac et al.,
All individual His6-tagged truncated 1996). The fourth day after this first
proteins (40 µg) were also used as overlay injection, they were microinjected again
on a leafhopper actin protein blot and with 0,2 µg of His6-tagged PGK or
interaction was revealed using anti-His truncations (PGK-FL4 or PGK-FL5) in
MAb (1:3,000) followed by peroxidase phosphate buffer (8 mM NaH2PO4, 2 mM
conjugate goat anti-mouse IgGs. For this Na2HPO4 [pH 7,4]). Injections with
far Western experiment, the blot of phosphate buffer and with a His6-tagged
leafhopper proteins was prepared as protein (subunit of Mycoplasma mycoïdes
follows: 20 µg of proteins issued from SC ATPase) were included as controls in
Ciha-1 cell culture were separated on 12,5 the experiment. Then, for each tagged
% SDS-PAGE and transferred to protein or control, re-injected insects were
nitrocellulose membrane. Then, the randomly divided in subgroups of 3 among
experiment was conducted in a manner Eppendorf® tubes on which a Parafilm®
similar to that described previously except membrane separated the leafhoppers from
that anti-His MAbs in place of of anti-actin SP4 medium as previously described
antibodies were used. (Foissac et al., 1996). When feeding
through the Parafilm® membrane, infected
Effect of recombinant PGK and its leafhoppers injected S. citri into the
truncations on S. citri attachment to and medium. After 24 h at room temperature,
entry into Ciha-1 cells the SP4 medium was collected and
Monolayers of Ciha-1 cells grown in 24- incubated at 32°C. After one week, the
well plates (approximately 2 x 105 cells per yellow colour in the tube, indicating the
well) were incubated with various growth of S. citri, was noticed and the
concentrations of S. citri His6-tagged PGK presence of spiroplasmas was verified with
and truncations PGK-FL4 and PGK-FL5. optical microscopic observations.
Cells without PGK treatment represent
positive control. Cells treated with BSA Statistical analyses
were also included as a control. Each assay For attachment to and invasion of S. citri
condition was carried out in triplicate. into Ciha-1 cells, Student’s t test was used.
After incubation with proteins for 2 h at For S.citri transmission assay, a chi-square
32°C, the cells were infected with a test (two-tailed) was performed to identify
suspension of S. citri at a multiplicity of significant differences.
infection (MOI) of 15 to 30. After
infection with S. citri, spiroplasma RESULTS
68](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-161-320.jpg)














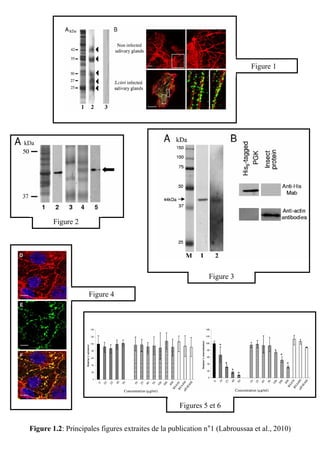
![A
B
SeqID: readseq.input(1), 412 bases, C98338A0 checksum.
Analysis Report:
CMSVM+ Unknown [No details]
CWSVM+ Unknown [No details]
CytoSVM+ Unknown [No details]
ECSVM+ Extracellular [No details]
ModHMM+ Unknown No internal helices found]
Motif+ Unknown [No motifs found]
Profile+ Unknown [No matches to profiles found]
SCL-BLAST+ Cytoplasmic [matched 3122608: Phosphoglycerate kinase]
SCL-BLASTe+ Unknown [No matches against database]
Signal+ Unknown [No signal peptide detected]
Localization Scores:
Cytoplasmic 5.86
CytoplasmicMembrane 0.00
Cellwall 0.30
Extracellular 3.83
Final Prediction:
Unknown (This protein may have multiple localization sites.)
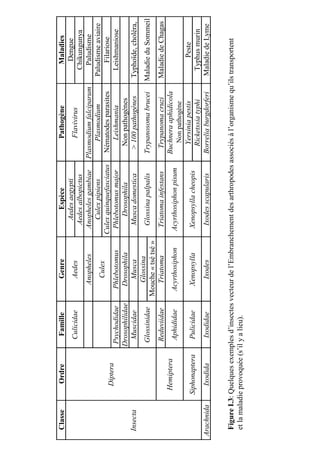
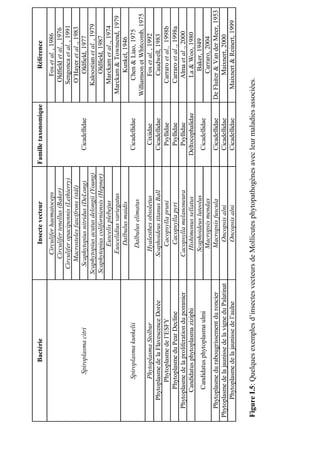
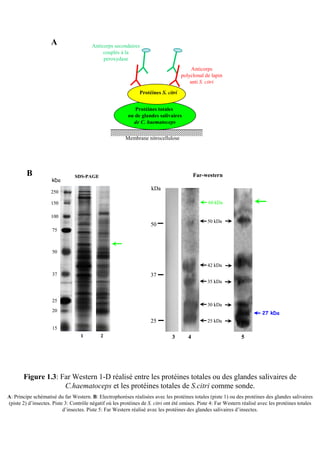
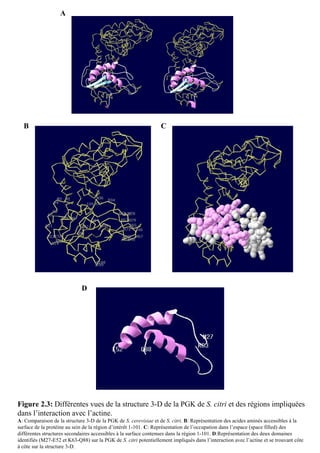
Figure 2.5: Localisation cellulaire de la PGK de S.citri
prédite par l’outil psortb v.3.0.2.
A: Interface d’interrogation avec la séquence de la PGK de S.citri et les quelques paramètres requis pour
cette interrogation. B: Résultats de l’interrogation avec les données importantes encadrées en rouge.](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-178-320.jpg)








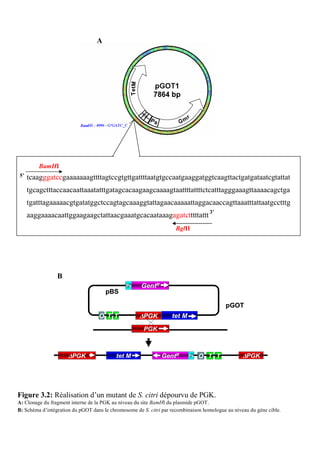

















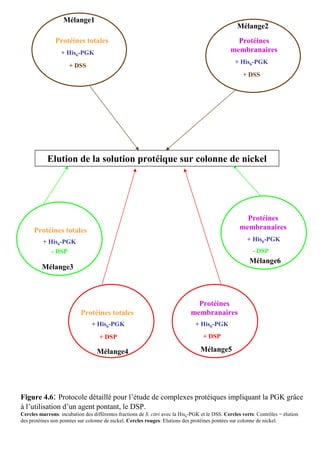

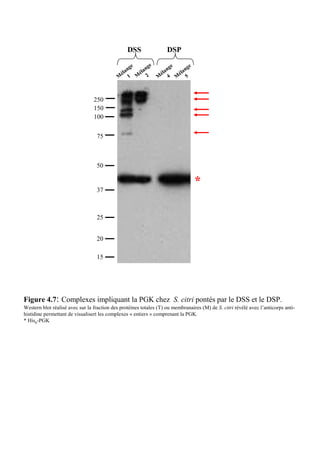

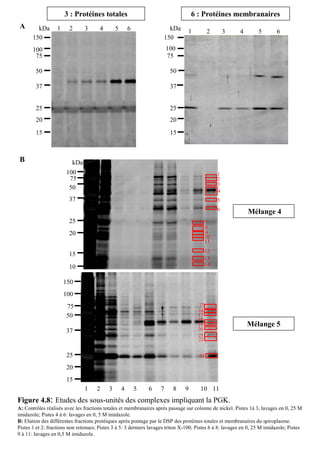

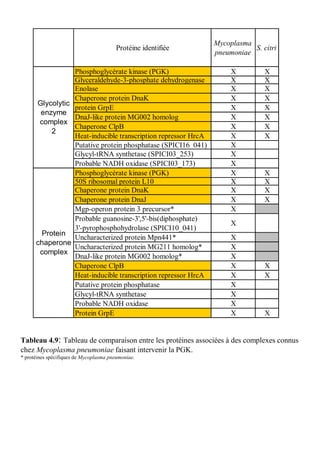


























![Matériels et méthodes
La souche DH10B [F- mcrA ∆(mrr-hsdRMS-mcrBC)Φ80lacZ∆M15 ∆lacX74 recA1
endA1 araD139 ∆(ara, leu)7697 galU galK λ- rpsL nupG] est couramment utilisée pour la
propagation de plasmides. Elle présente les avantages de posséder une grande efficacité de
transformation et la capacité à maintenir de façon stable des plasmides de grande taille.
La souche BL21 Star™ (DE3) a servi lors de la production des protéines et des
peptides recombinants. Elle permet d’améliorer le rendement de production dans un système
d’expression utilisant le promoteur T7 comme cela est le cas pour le vecteur d’expression
pET28a(+) (Novagen). En effet, elle porte une mutation dans le gène codant pour la RNaseE
(rne 131) qui est la principale source dégradation des ARNm.
Ces deux souches, après transformation, ont été cultivées en milieu LB (Tableau M.2)
solide à 37°C en présence d’ampicilline (100 µg/ml) ou de kanamycine (30 à 50 µg/ml).
En milieu liquide, la culture s’effectue à 37 °C sous agitation (180-200 rpm/min).
3. L’insecte vecteur : la cicadelle Circulifer haematoceps
3.1. Origine et conditions d’élevage
Les cicadelles adultes Circulifer haematoceps ont été prélevées sur Matthiola
sinuata en Corse. De retour au laboratoire, ces cicadelles ont été confinées quelques jours sur
giroflées (Matthiola incana). Dès la première ponte, les adultes ont été éliminés afin de
s’assurer que la population ne soit pas infectée par le spiroplasme. Aucune transmission
transovarienne n’ayant été démontrée chez S. citri à ce jour, les larves de ces insectes ne
présentent donc aucun risque d’être infectées par le spiroplasme. Les cicadelles saines sont
élevées dans des cages de plexiglas ventilées contenant trois giroflées et celles-ci sont
renouvelées une par une toutes les 2 semaines. La photopériode est de 16 heures à 30 °C ± 2
°C et 8 heures à 26 °C ± 2 °C. Les cages d’élevages sont regroupées dans une pièce
climatisée, isolées de l’extérieur par des filets pare-insectes et séparées des serres de
production de plantes par un sas de sécurité. Toutes les salles disposent d’un piège jaune
englué qui capte les insectes pouvant s’échapper lors de leur manipulation ou de l’entretien
des cages.
3.2. Capture et dissection des cicadelles
Les cicadelles sont capturées à l’aide d’un aspirateur à bouche muni d’un tube
collecteur. Elles sont anesthésiées sous gaz carbonique. Pour le prélèvement des glandes
110](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-251-320.jpg)











![Matériels et méthodes
sans agitation. 250 µl sont incubés sur milieu solide en présence de kanamycine (30 µg/ml) à
37 °C.
9.2. S. citri
Les spiroplasmes sont transformés par électroporation selon la méthode décrite par
McCammon (McCammon et al., 1990) et modifiée par Stamburski (Stamburski et al., 1990).
Trois millilitres d’une culture de S. citri sont centrifugés à 20 000 g pendant 15 minutes à 18
°C. Le culot de cellules est remis en suspension dans 2 ml de tampon Hepes-Saccharose (HS ;
8 mM Hepes, 280 mM Saccharose [pH 7,4]). Après une deuxième centrifugation dans les
mêmes conditions, le culot est finalement remis en suspension dans 400 µL de tampon HS.
Pour la transformation, 1 à 2 µg d’ADN plasmidique sont mélangés à 400 µl de
cellules dans une cuvette d’électroporation (distance inter-électrodes de 0,4 cm) et celle-ci est
maintenue dans la glace pendant 10 minutes. Un témoin sans ADN est systématiquement
réalisé afin de suivre l’apparition éventuelle de résistants spontanés. Les transformations sont
effectuées avec un électroporateur Gene Pulser® (BIO-RAD) réglé sur une tension de 6,25
kV/cm, une capacité de 3 µF et une résistance de 1 000 Ω. Dans ces conditions, la durée du
choc électrique est d’environ 1 à 3 ms. Après 3 heures d’expression phénotypique à 32 °C
dans 1 ml de milieu SP4, des dilutions 100, 10-1 et 10-2 du mélange de transformation sont
étalées sur milieu SP4 solide contenant de la gentamicine (100 µg/ml). Après 2 à 3 semaines
d’incubation à 32 °C, les colonies sont ensemencées en milieu liquide contenant l’antibiotique
adéquat. Ce premier ensemencement correspond au premier passage.
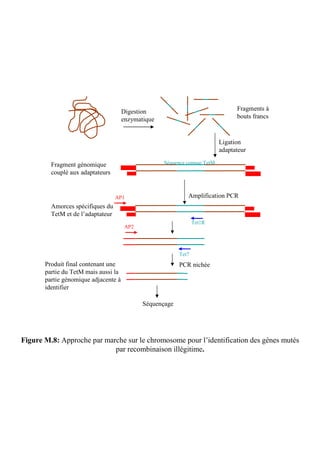
10. Marche sur le chromosome
La technique de marche sur le chromosome a été réalisée en utilisant le kit
GenomeWalker™ DNA walking (BD Biosciences) dont le principe est schématisé sur la
figure M.8.
2 µg d’ADN chromosomique de chacun des mutants de S. citri considéré ont été
digérés par une des deux endonucléases SmaI ou HincII pendant 4 h à 37°C. Le choix des
amorces tient compte de deux paramètres importants qui sont, dans un premier temps, que les
enzymes doivent digérer l’ADN donnant des bouts francs afin de permettre l’ajout par ligation
d’adaptateurs indispensables aux amplifications ultérieures. Dans un second temps, les
enzymes ne doivent pas coupées avec une fréquence élevée afin de ne pas digérer l’ADN dans
la région d’intérêt.
118](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-267-320.jpg)


































![Matériels et méthodes
série de dilutions au 1/10ème est réalisée à partir du filtrat. Après incubation à 32°C, le
changement de couleur est noté et le nombre de spiroplasmes est déterminé.
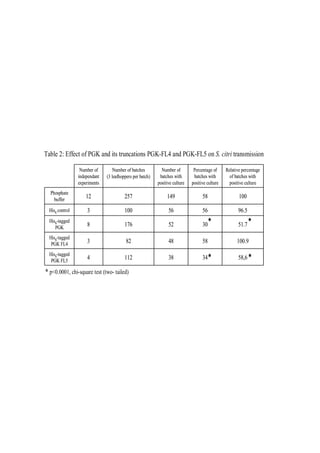
3. Effet de la PGK et des peptides sur l’adhésion et/ou l’internalisation
3.1. Adhésion
Les cellules Ciha-1, cultivées dans des plaques 24 puits (environ 105 cellules par puit),
sont incubées pendant 2 h à 32°C dans 900 µl de milieu cicadelle additionné de différentes
concentrations de PGK de Saccharomyces cerevisiae (Sigma), de His6-PGK ou de His6-
PGKFL4 et FL5. Les cellules n’ayant subi aucun traitement servent de contrôles positifs tandis
que les cellules non infectées par S. citri servent de contrôles négatifs. Des concentrations
équivalentes de BSA ou de His6-eIF4E servent également de contrôles. Chaque condition est
réalisée en triplicat au cours de chacune des expériences indépendantes menées.
Après cette incubation, les cellules sont infectées avec 100 µl d’une culture de S. citri
avec une multiplicité d’infection (MOI) d’environ 15-30 pendant 4 h à 4°C. Cette température
permet aux spiroplasmes d’adhérer aux cellules de l’hôte mais inhibe les processus eucaryotes
mis en place au cours de l’internalisation. Les cellules sont ensuite lavées dans 500 µl de
milieu de Schneider (Lonza) afin d’éliminer les spiroplasmes libres. Les cellules sont
trypsinées 10 min à 32°C avec un tampon TrypLE™ (Invitrogen) puis des dilutions des
cellules, sur lesquelles des spiroplasmes ont adhérés, sont réalisées. Le traitement avec cette
solution de trypsine n’a aucun effet sur les spiroplasmes (Duret et al., 2009). Ces différentes
dilutions sont étalées sur des boites contenant le milieu de culture des spiroplasmes. Après un
temps d’incubation d’environ une semaine à 32°C, le nombre de colonies est dénombré afin de
déterminer le nombre de cellules sur lesquelles au moins un spiroplasme a adhéré. Un
pourcentage relatif d’adhésion des spiroplasmes sur les cellules Ciha-1 est calculé comme suit:
[(Nombre de colonies dénombrées avec incubation protéine exogène / nombre de colonies
dénombrées sans traitement) x 100%]. Un test de Student a été réalisé afin de déterminer
l’indice de confiance des résultats obtenus.
3.2. Internalisation
L’effet du traitement par les différents peptides de la PGK sur l’internalisation des
spiroplasmes dans les cellules Ciha-1 a été déterminé par un test de protection à la
gentamycine (Isberg & Falkow, 1985). La méthodologie utilisée est identique à celle mise au
138](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-307-320.jpg)

























![Bibliographie
Gardy, J. L., Laird, M. R., Chen, F., Rey, S., Walsh, C. J., Ester, M. and Brinkman, F. S.
(2005). "PSORTb v.2.0: expanded prediction of bacterial protein subcellular
localization and insights gained from comparative proteome analysis." Bioinformatics
21(5): 617-23.
Garnier, M., Clerc, M. and Bove, J. M. (1984). "Growth and division of Spiroplasma citri:
elongation of elementary helices." J Bacteriol 158(1): 23-8.
Garnier, M., Foissac, X., Gaurivaud, P., Laigret, F., Renaudin, J., Saillard, C. and Bove, J. M.
(2001). "Mycoplasmas, plants, insect vectors: a matrimonial triangle." C.R.
Acad.Sci.Paris. 324(10): 923-8.
Gaurivaud, P., Danet, J. L., Laigret, F., Garnier, M. and Bove, J. M. (2000). "Fructose
utilization and phytopathogenicity of Spiroplasma citri." Mol Plant Microbe Interact
13(10): 1145-55.
Gaurivaud, P., Laigret, F., Garnier, M. and Bove, J. M. (2000). "Fructose utilization and
pathogenicity of Spiroplasma citri: characterization of the fructose operon." Gene
252(1-2): 61-9.
Gaurivaud, P., Laigret, F., Garnier, M. and Bove, J. M. (2001). "Characterization of FruR as a
putative activator of the fructose operon of Spiroplasma citri." FEMS Microbiol Lett
198(1): 73-8.
Gaurivaud, P., Laigret, F., Verdin, E., Garnier, M. and Bove, J. M. (2000). "Fructose operon
mutants of Spiroplasma citri." Microbiology 146 ( Pt 9): 2229-36.
Gerdes, K., Moller-Jensen, J. and Bugge Jensen, R. (2000). "Plasmid and chromosome
partitioning: surprises from phylogeny." Mol Microbiol 37(3): 455-66.
Ghosh, A. K., Devenport, M., Jethwaney, D., Kalume, D. E., Pandey, A., Anderson, V. E.,
Sultan, A. A., Kumar, N. and Jacobs-Lorena, M. (2009). "Malaria parasite invasion of
the mosquito salivary gland requires interaction between the Plasmodium TRAP and
the Anopheles saglin proteins." PLoS Pathog 5(1): e1000265.
Ghosh, A. K., Ribolla, P. E. and Jacobs-Lorena, M. (2001). "Targeting Plasmodium ligands
on mosquito salivary glands and midgut with a phage display peptide library." Proc
Natl Acad Sci U S A 98(23): 13278-81.
Gianfaldoni, C., Censini, S., Hilleringmann, M., Moschioni, M., Facciotti, C., Pansegrau, W.,
Masignani, V., Covacci, A., Rappuoli, R., Barocchi, M. A. and Ruggiero, P. (2007).
"Streptococcus pneumoniae pilus subunits protect mice against lethal challenge."
Infect Immun 75(2): 1059-62.
Gildow, F. E. (1987). "Virus-membrane interactions involved in circulative transmission of
luteoviruses by aphids " Adv. Dis. Vector Res. 4: 93-120.
Giron, J. A., Lange, M. and Baseman, J. B. (1996). "Adherence, fibronectin binding, and
induction of cytoskeleton reorganization in cultured human cells by Mycoplasma
penetrans." Infect Immun 64(1): 197-208.
Glew, M. D., Marenda, M., Rosengarten, R. and Citti, C. (2002). "Surface diversity in
Mycoplasma agalactiae is driven by site-specific DNA inversions within the vpma
multigene locus." Journal of Bacteriology 184(21): 5987-5998.
Gouin, E., Egile, C., Dehoux, P., Villiers, V., Adams, J., Gertler, F., Li, R. and Cossart, P.
(2004). "The RickA protein of Rickettsia conorii activates the Arp2/3 complex."
Nature 427(6973): 457-61.
Gouin, E., Welch, M. D. and Cossart, P. (2005). "Actin-based motility of intracellular
pathogens." Curr Opin Microbiol 8(1): 35-45.
Gouranton, J. and Folliot, R. (1970). "[Presence of perinuclear microtubules in the salivary
gland of homopterous insect. Centrotus cornutus L]." C R Acad Sci Hebd Seances
Acad Sci D 270(14): 1819-21.
Granett, A. L., Blue, R. L., Harjung, M. K., Calavan, E. C. and Gumpf, D. J. (1976).
"Occurence of Spiroplasma citri in periwinkle in California." Calif. Agric. 30: 18-19.
Grassme, H. U. C., Ireland, R. M. and VanPutten, J. P. M. (1996). "Gonococcal opacity
protein promotes bacterial entry-associated rearrangements of the epithelial cell actin
cytoskeleton." Infection and Immunity 64(5): 1621-1630.
147](https://image.slidesharecdn.com/labroussaa2-120306174735-phpapp01/85/Labroussaa-2-337-320.jpg)




























































