Le document traite des mécanismes d'adsorption d'atomes et de molécules sur la surface Si(001)-(2×1) en utilisant des techniques de spectroscopie de photons et d'électrons. Il aborde les structures, la préparation, les propriétés optiques de la surface, ainsi que l'interaction avec différentes molécules simples et organiques. Enfin, il présente des projets de recherche futurs et des méthodologies pour étudier ces phénomènes.










![TABLE DES MATI`ERES vii
vicinal par spectroscopies optiques et microscopie `a effet tunnel. J’ai notamment ´etudi´e l’adsorption
d’´ethyl`ene et des calculs DFT effectu´es `a Rome dans l’´equipe de Rodolfo Del Sole, ont donn´es de tr`es
bons r´esultats qui seront d´etaill´es dans ce manuscrit et qui permettent dans ce cas d’obtenir des infor-
mations sur la g´eom´etrie d’adsorption de l’´ethyl`ene pour diff´erents taux de couverture. J’ai ´egalement
particip´e `a l’´etude de l’adsorption de ph´enylac´etyl`ene/Si(001)-(2×1) principalement effectu´ee par avec
Olivier Pluchery, Maˆıtre de Conf´erences dans l’´equipe depuis septembre 2002 et Romain Coustel.
L’´etude conjointe par STM et RAS des surfaces vicinales de silicium nous a permis de montrer que
la RAS permet d’estimer la proportion des deux domaines orthogonaux pr´esents sur la surface. L’in-
conv´enient des surfaces vicinales de silicium est qu’elles comportent un grand nombre de marches
qui contribuent notablement au signal de r´eflectivit´e anisotrope. Afin de pouvoir travailler sur des
surfaces monodomaines, mais sans marches, j’ai mis au point un protocole exp´erimental permettant
la pr´eparation de surfaces nominales de Si(100)-(2×1) monodomaine par ´electromigration `a haute
temp´erature. L’obtention de cette surface n´ecessite une tr`es grande vigilance quant aux conditions
exp´erimentales, ainsi nous sommes la troisi`eme ´equipe `a publier le spectre de r´eflectivit´e anisotrope
d’une surface de silicium nominale. Je me suis alors attach´ee `a pr´eparer ces surfaces et `a ´etudier
l’adsorption d’hydrog`ene et d’oxyg`ene en spectroscopie optique de surface. Nous travaillons en col-
laboration avec des ´equipes de th´eoriciens ( Rodolfo Del Sole de Rome, Giovanni Onida de Milan,
et Friedhelm Bechstedt de Jena) qui ont pu calculer les spectres de r´eflectivit´e anisotrope et/ou de
r´eflectivit´e diff´erentielle de l’hydrog`ene et de l’oxyg`ene adsorb´es sur Si(100)-(2×1) (articles 1, 2).
Transition de phase dans les films minces MnAs/GaAs(001)
Depuis notre arriv´ee `a Boucicaut, j’ai ´egalement eu l’occasion de collaborer avec le projet “Couches
minces et nanostructures hybrides” de l’INSP pour ´etudier par spectroscopies optiques de surface la
transition de phase qui existe au environ de 50◦C dans les films minces MnAs/GaAs(001) pr´epar´es
par ´evaporation sous vide `a Jussieu. Les mesures de RAS ont principalement ´et´e effectu´ees par Olivier
Pluchery, Frank Vidal et Y. Borensztein, ex situ. Le compos´e MnAs ´epitaxi´e sur GaAs(100) pr´esente
une phase α hexagonale et une phase β orthorhombiques qui co-existent entre 10◦C et 45◦C. Alors
que dans le compos´e MnAs la phase β est paramagn´etique alors que dans le syst`eme en couche
mince ´epitaxi´ees MnAs/GaAs(001), il semble qu’elle soit plutˆot antiferromagn´etique. Des mesures
STM effectu´ees in situ `a l’INSP ont montr´e que les deux phases du compos´e ´epitaxi´e ne se m´elangent
pas mais s’auto organisent en formant des bandes alternativement de phase α et β le long de la direction
[001]. Comme les deux phases α et β pr´esentent une anisotropie cristalline ainsi qu’une structure
´electronique diff´erente, nous avons voulu suivre leur transition de phase par RAS. Les mesures ont
montr´e des spectres distincts pour la phase β obtenue `a 50◦ et la phase α obtenue autour de 5◦C
provenant essentiellement d’une anisotropie volumique dans les deux phases. Dans ces conditions,
il est possible de reproduire les spectres interm´ediaires en effectuant une combinaison lin´eaire des
spectres correspondants aux deux phases et d’en d´eduire l’abondance relative des deux phases durant
la transition. Les r´esultats sont coh´erents avec ceux obtenus par diffraction des rayons X et d´emontrent
que la RAS permet d’´etudier cette transition de mani`ere ais´ee (article 3).
Nanocatalyse
En 2002 - 2003, notre ´equipe en collaboration avec des chimistes du Laboratoire de R´eactivit´e
des Surfaces (LRS) de l’UPMC ainsi que d’autres laboratoires `a Grenoble et Marseille, a obtenu des
financements universitaire (BQR) et national (ACI) sur un projet concernant l’´etude de la nanocatalyse
sur des particules d’or. En effet, alors que l’effet catalytique de l’or volumique est pratiquement nul
`a temp´erature, il pr´esente une activit´e catalytique importante sous forme de nanoparticules d’environ
2-5 nm de diam`etre. L’objectif de ce projet ´etait l’´elaboration de ces nanoparticules d’or sur diff´erents](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-11-320.jpg)











![Chapitre 2
Techniques exp´erimentales
2.1 Les spectroscopies de photons
2.1.1 La spectroscopie de r´eflectivit´e anisotrope ou RAS
Principe
La spectroscopie de r´eflectivit´e anisotrope (acronyme anglais RAS pour Reflectance Anisotropy
Spectroscopy) ´egalement appel´ee spectroscopie de r´eflectivit´e diff´erentielle (acronyme anglais RDS
pour Reflectance Difference Spectroscopy), est une technique optique originale qui a ´et´e d´evelopp´ee
par D.E. Aspnes [1] pour suivre, en temps r´eel, la croissance de semi-conducteurs de type III-V. Par
cette technique, analogue `a l’ellipsom´etrie en incidence normale, des photons polaris´es parall`element
ou perpendiculairement aux axes principaux d’un ´echantillon sont envoy´es sous incidence normale. On
s’int´eresse alors `a mesurer la quantit´e ∆r
r , qui repr´esente la diff´erence de r´eflectivit´e normalis´ee de la
surface :
∆r
r
=
rx − ry
r
= −
2ω
c
i
x
s − y
s
volume − 1
(2.1)
o`u rx et ry sont les r´eflectivit´es selon les axes principaux de la surface et o`u x
s , y
s et volume sont
respectivement les tenseurs di´electriques de surface et de volume. Comme les tenseurs di´electriques de
surface d´ependent des transitions ´electroniques entre ´etats pleins et ´etats vides, cette technique peut
ˆetre utilis´ee pour ´etudier les propri´et´es ´electroniques des surfaces. Toutefois, compte tenu du nombre
important d’´etats qui peuvent intervenir dans les transitions, une interpr´etation directe des spectres
est difficile et le recours `a des calculs th´eoriques ab initio, comme on le verra par la suite, s’av`ere
indispensable pour pouvoir interpr´eter correctement les r´esultats obtenus.
Malgr´e la tr`es grande profondeur de p´en´etration de la lumi`ere dans la mati`ere, l’acc`es aux propri´et´es
optiques de la surface est r´ealis´e en utilisant des ´echantillons qui poss`edent un volume isotrope et
une surface anisotrope comme dans le cas des surfaces de type (110) ou des surfaces reconstruites. La
contribution provenant du volume va s’annuler du fait de l’isotropie de celui-ci et la contribution de la
surface se retrouve ainsi exacerb´ee. La limitation de cette technique est de ne pouvoir ˆetre utilis´ee que
sur des ´echantillons pr´esentant une anisotropie en surface. Le signal RAS ´etant assez faible (quelques
dixi`emes de pourcent), il est pr´ef´erable d’utiliser un dispositif analogue `a celui pr´esent´e ci-dessous pour
augmenter la sensibilit´e de la technique. Le dispositif exp´erimental permettant ce genre de mesure
est pr´esent´e sur la figure 2.1. La lumi`ere incidente est polaris´ee lin´eairement et verticalement, elle
est ensuite r´efl´echie par l’´echantillon dont les axes principaux sont plac´es `a 45◦ de la direction de
polarisation. La lumi`ere traverse ensuite un modulateur photo-´elastique qui va produire un d´ephasage
entre l’axe vertical et l’axe horizontal `a une fr´equence de 50kHz. Le modulateur permet de r´eduire le
3](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-23-320.jpg)

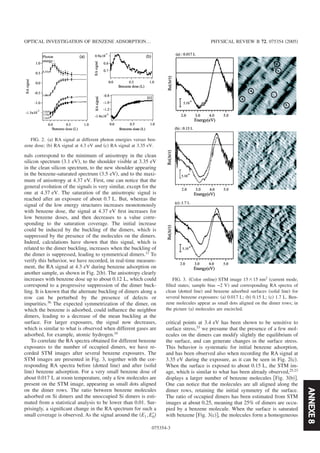
![2.1. LES SPECTROSCOPIES DE PHOTONS 5
x
yz
R0 R
(a) (b)
Fig. 2.2 — Principe de la SRDS. (a) : r´eflectivit´e de la surface nue R0, (b) r´eflectivit´e de la surface
avec adsorbats R .
En d´etectant `a la fr´equence de modulation ou `a deux fois la fr´equence de modulation on peut alors
avoir acc`es `a la partie imaginaire ou la partie r´eelle de la quantit´e ∆r
r .
Le dispositif exp´erimental, repr´esent´e sur la figure 2.1, se compose d’une lampe Xe, d’un polariseur,
d’un modulateur photo-´elastique, d’un analyseur, d’une s´erie de miroirs plans et paraboliques. La
lumi`ere est alors dispers´ee sur des r´eseaux plans en fonction de l’´energie des photons, puis l’acquisition
du signal se fait grˆace `a un multiplicateur de photo´electrons (PEM). La gamme ´energ´etique accessible
par cette technique va de 1.35 eV jusque 5.5 eV (ou de 918 nm `a 225 nm).
2.1.2 La spectroscopie de r´eflectivit´e diff´erentielle de surface ou SDRS
Principe
Cette technique mesure la diff´erence de r´eflectivit´e d’une surface lorsque celle-ci subit des modifi-
cations (adsorption d’atomes ou mol´ecules, d´esorption, variation de temp´erature...). La r´eflectivit´e de
la surface propre, R0, est enregistr´ee ensuite, la surface subit des modifications provoqu´ees par l’ad-
sorption d’atomes par exemple, entraˆınant une modification de la r´eflectivit´e not´ee R. Le dispositif
donne alors acc`es `a la quantit´e ∆R
R = R−R0
R0
sur une gamme d’´energie allant de 1.5 eV `a 5.5 eV (voir
figure 2.2). Cette technique peut ˆetre utilis´ee sur tout type de surface r´efl´echissante contrairement `a
la technique RAS qui n´ecessite des surfaces anisotropes. La dynamique du syst`eme ainsi que sa sensi-
bilit´e, permettent d’enregistrer un spectre complet toutes les deux secondes avec un rapport signal sur
bruit satisfaisant. Cette technique est id´eale pour ´etudier les cin´etiques d’adsorption qui ont lieu sur
des ´echelles de temps compatibles avec le dispositif. Toutefois, pour obtenir un signal interpr´etable, il
est imp´eratif d’avoir une tr`es bonne stabilit´e du syst`eme et de pouvoir s’affranchir du maximum de
vibrations. En effet, `a chaque instant, la r´eflectivit´e de l’´echantillon est compar´ee `a une r´eflectivit´e de
r´ef´erence et aucune perturbation ne doit avoir eu lieu entre les deux mesures pour qu’elles puissent
ˆetre fiables. Comme on peut le voir sur le sch´ema de la figure 2.3, le dispositif se compose d’une lampe
deut´erium, d’un polariseur permettant d’avoir la lumi`ere polaris´ee s ou p, d’une s´erie de lentilles et de
miroirs. La lumi`ere est ensuite dispers´ee par un prisme et envoy´ee sur une barrette de photodiodes,
l’acquisition se fait par l’interm´ediaire d’un analyseur optique multi canal refroidi par effet Peltier
pour plus de stabilit´e. Il est possible de montrer que la quantit´e ∆R
R mesur´ee sous incidence normale,
peut se mettre sous la forme suivante [2] :
∆R
R
=
8πω
c
Im
α(ω)
b − 1
(2.7)](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-25-320.jpg)



![2.3. DISPOSITIFS EXP´ERIMENTAUX 9
photo´emission `a haute r´esolution et des mesures d’absorption X, elle dispose ´egalement d’un d´etecteur
de fluorescence X utilis´e pour la diffusion in´elastique des photons. Afin de pouvoir ´etudier les diff´erentes
orientations de la polarisation des rayons X, la chambre d’analyse dispose d’un syst`eme permettant
d’effectuer des rotations autour de l’axe du faisceau. Plus de d´etails sur les caract´eristiques techniques
de la ligne I511 peuvent ˆetre obtenus dans l’article de Denecke et al. [3].](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-29-320.jpg)


![12 CHAPITRE 3. LA SURFACE SI(001)-(2×1)
r´eactions de cycloadditions en chimie organique, peuvent ˆetre observ´ees dans le cas de l’adsorption de
mol´ecules organiques poss´edant une double liaison.
En raison de la reconstruction (2×1) de la surface, celle-ci pr´esente un ´etat de surface d´elocalis´e le long
des rang´ees de dim`eres, faisant intervenir les ´etats de type π et π des liaisons pendantes ; cet ´etat de
surface a pu ˆetre mis en ´evidence d`es le tout d´ebut des ann´ees 90 par photo´emission [4]. R´ecemment,
des calculs de structure de bande de type DFT-LDA de la surface ont pu faire apparaˆıtre une diff´erence
d’´energie d’environ 1.1 eV [5] entre ´etats pleins et ´etats vides. Exp´erimentalement, l’´ecart entre les
´etats π et π des liaisons pendantes se situe `a environ 1.6 eV et est donc accessible aux techniques
optiques utilis´ees `a l’INSP [6]. Cet ´etat de surface est tr`es sensible `a la qualit´e de la reconstruction de
la surface et aux adsorptions comme on le verra dans la suite.
3.2 Pr´eparation
3.2.1 Surface nominale
Lorsqu’une surface nominale est pr´epar´ee sous ultra haut vide par chauffage, il se forme des ter-
rasses monoatomiques o`u les rang´ees de dim`eres sont orient´ees `a 90◦ les unes des autres pour minimiser
l’´energie de surface lors du refroidissement. Sur ce type de surface, il existe en moyenne autant de
rang´ees dans un sens que dans l’autre, la rendant ainsi isotrope et impossible `a ´etudier en utilisant
la technique RAS. Pour rem´edier `a cet inconv´enient, il est possible de rendre la surface anisotrope en
faisant migrer les marches et obtenir ainsi de tr`es larges domaines o`u toutes les rang´ees de dim`eres
sont orient´ees dans la mˆeme direction. L’´electromigration peut ˆetre utilis´ee pour pr´eparer ce type de
surface. En effet, en faisant passer un courant au travers d’un ´echantillon parfaitement orient´e (de
d´esorientation maximale de 0.05◦) et port´e `a haute temp´erature (sup´erieure `a 1000◦C), les marches
deviennent mobiles et s’accumulent sur un cˆot´e de l’´echantillon [7]. Afin d’obtenir une bonne qualit´e
de surface, il est imp´eratif de garder la pression dans la gamme des 10−10 T pendant toute l’op´eration
car sinon du carbone va s’incorporer dans le silicium et diminuer la mobilit´e des marches. Ainsi, pour
r´eussir l’´electromigration, il faut ˆetre capable de pouvoir chauffer le silicium par passage de courant
direct en le portant `a une temp´erature sup´erieure `a 1000◦C pendant au moins une heure tout en gar-
dant un vide de tr`es bonne qualit´e. Les nombreuses exp´eriences, que j’ai men´ees, ont montr´e qu’il faut
au minimum une journ´ee enti`ere pour r´ealiser l’´electromigration et obtenir un spectre de r´eflectivit´e
anisotrope de grande amplitude.
3.2.2 Surface vicinale
Une autre alternative permet d’obtenir des surfaces monodomaines en utilisant des ´echantillons de
silicium vicinaux d´esorient´es de 4◦ ou 5◦. Lorsqu’un ´echantillon de ce type est port´e `a 1000◦C pendant
une dizaine de secondes dans une pression n’exc´edant pas quelques 10−10 T, les marches s’apparient
pour former des doubles marches. Sur les terrasses, qui comportent environ 6 `a 8 dim`eres, toutes
les rang´ees sont orient´ees perpendiculairement aux doubles-marches, rendant la surface anisotrope `a
l’´echelle macroscopique. Cette surface est facile et rapide `a pr´eparer, l’´echantillon est mis une nuit en
d´egazage `a 650◦C et ensuite port´e `a 1050◦C pendant 10 `a 20 secondes pour retirer la couche d’oxyde
natif et obtenir la reconstruction soit par passage de courant direct, comme c’est le cas `a l’INSP, soit
par bombardement ´electronique, cette derni`ere proc´edure a ´et´e utilis´ee avec succ`es lors des mesures
sur la ligne synchrotron I511. Aucune proc´edure sp´ecifique n’est utilis´ee au cours du refroidissement
de l’´echantillon, le crit`ere d´eterminant la bonne qualit´e de la surface ´etant la pression qui ne doit
jamais exc´eder quelques 10−10 T pendant les chauffages [8]. L’exp´erience a montr´e qu’un minimum](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-32-320.jpg)
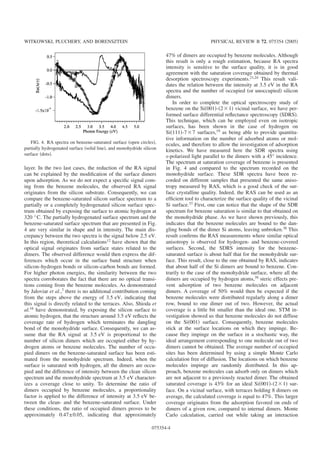
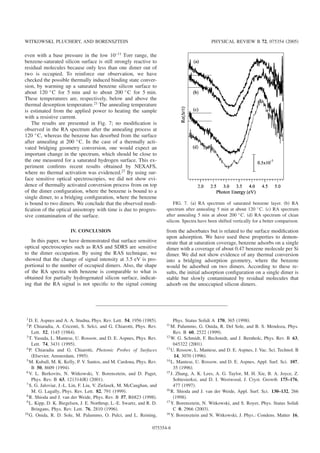
![3.3. PROPRI´ET´ES OPTIQUES 13
(b)
-3
-2
-1
0
1
-3
-2
-1
0
1
5.55.04.54.03.53.02.52.01.5
Energie (eV)
(d)
E2E1 E'0
Re(∆r/r)x10
-3
Re(∆r/r)x10
-3
200x200 nm2
100x100 nm2
(a)
(c)
Fig. 3.2 — (a) : Image STM 200×200 nm2 d’une surface nominale apr`es ´electromigration, (b) : spectre
de RA sur une surface nominale monodomaine, (c) : Image STM 100×100 nm2 d’une surface vicinale
4◦, (d) : Spectre RA de d’une surface vicinale 4◦. Structure de bande calcul´ee du Si en volumr et en
surface (001) (d’apr`es [5]).
de trois chauffages est n´ecessaire pour obtenir une surface de qualit´e standard, observable par STM,
poss`edant une densit´e de d´efaut de moins que 5%.
3.3 Propri´et´es optiques
3.3.1 Surfaces monodomaines
La surface de silicium (001) est ´etudi´ee par la technique RAS depuis une dizaine d’ann´ees, autant
exp´erimentalement que th´eoriquement, toutefois, l’origine de toutes les structures pr´esentes dans les
spectres n’est pas encore bien comprise. La figure 3.2 pr´esente les spectres de r´eflectivit´e anisotrope
d’une surface nominale obtenue apr`es ´electromigration des marches et d’une surface vicinale d´esorient´ee
de 4◦, pr´epar´ee en chauffant par passage de courant direct. Les deux lignes verticales indiquent les
positions des points critiques E2 et E1, E0. Bien que la RAS soit une technique qui permette de sonder
les surfaces anisotropes en annihilant la contribution provenant du volume isotrope, au voisinage de
la surface, les ´etats ´electroniques de volume sont perturb´es et deviennent `a leur tour anisotropes
compliquant la compr´ehension des spectres. L’origine des diff´erentes structures peut ˆetre ´elucid´ee
grˆace `a des calculs ab initio de type DFT-LDA. Plusieurs reconstructions de la surface (001) ont ´et´e
test´ees et il semble que le meilleur accord avec l’exp´erience soit obtenu pour une reconstruction de type
c(4×2) o`u les dim`eres pr´esentent une asym´etrie [9, 10]. Ainsi, la structure positive `a 4.4 eV correspond
aux transitions optiques provenant d’´etats ´electroniques de volume modifi´es par la surface, qui ont lieu
au point critique X de la zone de Brillouin. Il en va de mˆeme pour la structure positive `a 3.2 eV qui
correspond aux transitions de volume modifi´ees par la surface aux points critiques L et Γ [10, 11]. Alors
que cette derni`ere structure est bien marqu´ee dans le cas de la surface nominale, elle apparaˆıt comme
un ´epaulement dans le spectre mesur´e sur une surface vicinale. Cette structure semble tr`es sensible
aux contraintes de surface provoqu´ees par la pr´esence de dim`eres et/ou de marches comme il a ´et´e
montr´e par Hingerl et al. [11]. D’autre part, les calculs ab initio r´ealis´es par Schmidt et al., indiquent](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-33-320.jpg)
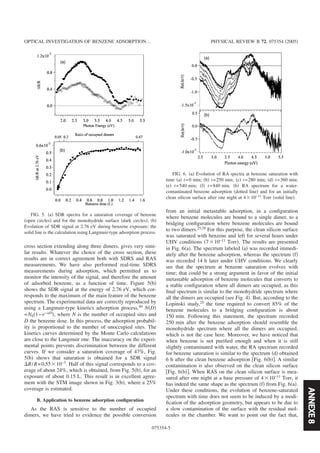
![14 CHAPITRE 3. LA SURFACE SI(001)-(2×1)
(a) (c)
4nm4nm
-4x10
-3
-3
-2
-1
0
1
Re(∆r/r)
5.55.04.54.03.53.02.52.01.5
Energie de photon (eV)
(b)
Fig. 3.3 — (a) : Image STM 100×100 nm2 d’une surface vicinale apr`es quelques flashes pour ´eliminer la
couche d’oxyde, (b) : Spectre de RA d’une surface fraˆıchement flash´ee (en rouge), de la mˆeme surface
apr`es de tr`es nombreux flashes `a 1050◦C (en bleu) et de la surface vicinale id´eale (en vert), (c) : Image
STM 100×100 nm2 de la surface vicinale 4◦ apr`es de tr`es nombreux flashes.
´egalement qu’il n’y a pas de transitions provenant d’´etats du volume pour des ´energies inf´erieures `a
3.2 eV [10]. Ainsi, la structure n´egative qui est visible `a 1.6 eV sur le spectre de RA de la surface
nominale provient de transitions de l’´etat de surface d´elocalis´e le long de la rang´ee de dim`eres [10, 9].
En revanche, on devine `a peine cette structure sur le spectre de la surface vicinale, en effet, il est
possible que la pr´esence des marches provoque une modification de la structure de bande de cet ´etat
de surface et entraˆıne un ´elargissement de la structure correspondante. Vers 3 eV, on note ´egalement
la pr´esence d’une structure n´egative et assez large dans le cas de la surface nominale. L’origine de cette
structure n’a pas ´et´e clairement mise en ´evidence, mais il semble ´etabli, par des exp´eriences impliquant
des adsorptions de mol´ecules, qu’elle est relative `a la pr´esence des liaisons pendantes sur les dim`eres
de silicium [12]. L’´etude des spectres de r´eflectivit´e anisotrope sur les surfaces vicinales est compliqu´e
par la pr´esence de marches, comme le montre l’image STM de la figure 3.2(c). Pratiquement toute
la surface de l’´echantillon est couverte de terrasses s´epar´ees par des doubles marches orient´ees dans
la mˆeme direction. Le spectre de RA pr´esente, vers 3.1 eV, une structure associ´ee `a la pr´esence des
marches. Cette structure est d’autant plus marqu´ee que la vicinalit´e de la surface est grande [13, 6].
Toutefois, il a ´et´e montr´e qu’au dessus de 3.4 eV, l’influence des marches devient n´egligeable et donc
que le signal de RA provient essentiellement des terrasses [13].
3.3.2 Quantification du m´elange des deux domaines
La RAS est un outil id´eal pour caract´eriser le m´elange des deux domaines sur une surface de
silicium. En effet, sur une surface nominale standard obtenue apr`es avoir retir´e la couche d’oxyde
`a 1000◦C environ, celle-ci pr´esente un m´elange de deux domaines o`u les dim`eres sont orient´es `a
90◦ les uns des autres. Sur une telle surface le signal de RA d’un des domaines annule le signal
de RA de l’autre domaine, rendant la surface isotrope `a l’´echelle macroscopique. Pour obtenir une
surface monodomaine, on peut utiliser des surfaces vicinales qui, apr`es avoir ´et´e pr´epar´ees par la
proc´edure d´ecrite pr´ec´edemment, peuvent n´eanmoins pr´esenter une proportion non n´egligeable de
dim`eres orient´es perpendiculairement `a l’orientation majoritaire des dim`eres sur la surface. Ainsi, la
technique de RAS informe directement sur la proportion des domaines qui coexistent `a la surface
de l’´echantillon vicinal sens´e ˆetre monodomaine. Pour obtenir cette information, il a fallu ´etalonner
le signal de RA de la surface vicinale en fonction de la topographie r´eelle de la surface. Cela est](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-34-320.jpg)
![3.3. PROPRI´ET´ES OPTIQUES 15
-3x10
-3
-2
-1
0
1
Re(∆r/r)
5.55.04.54.03.53.02.52.01.5
Energie de photon (eV)
-3x10
-3
-2
-1
0
1
Re(∆r/r)
5.55.04.54.03.53.02.52.01.5
Energie de photon (eV)
(a) (b)
Fig. 3.4 — (a) : Spectre de RA enregistr´e sur une surface Si(001) vicinale 2◦ (cercles orange) et spectre
simul´e en consid´erant 24 % de la surface occup´ee par le second domaine (ligne verte) ; (b) : Spectre
de RA enregistr´e sur une surface Si(001) vicinale 4◦ d´et´erior´ee par de nombreux flashes (cercles bleu)
et spectre simul´e en consid´erant 23 % de la surface occup´ee par le second domaine (ligne verte)
possible en analysant les images donn´ees par STM avec les spectres de RA correspondants. La figure
3.3(a), donne un exemple de la topographie de la surface juste apr`es avoir retir´e la couche d’oxyde par
chauffage rapide. Le signal de RA correspondant `a cette surface est montr´e sur la figure 3.3(b) en rouge
et l’anisotropie totale atteint alors 4.2×10−3. Lorsque la surface est re-pr´epar´ee de nombreuses fois, les
marches, qui sont nombreuses sur les surfaces vicinales, s’accumulent sur des d´efauts probablement dus
`a du carbure de silicium incorpor´e lors du recuit. La surface pr´esente alors de nombreuses protub´erances
ayant jusque 10 nm de hauteur et repr´esentant jusque 15 % de la surface totale, comme on le voit sur
la figure 3.3(c). Au pied des protub´erances, on distingue la pr´esence de deux domaines o`u les dim`eres
ont une orientation diff´erente. Le spectre de RA correspondant `a cette surface est pr´esent´e sur la figure
3.3(b) en bleu, il pr´esente une anisotropie totale beaucoup plus faible atteignant seulement 2.5×10−3.
Afin de caract´eriser la qualit´e de la surface `a l’aide de la RAS, il est int´eressant d’essayer de connaˆıtre
le signal de RAS d’une surface vicinale id´eale sans d´efauts. Cela est possible en corr´elant les images de
la topographie par STM et le spectre de RA mesur´e sur la mˆeme surface fraˆıchement flash´ee. Ainsi,
une ´etude statistique r´ealis´ee sur une surface de 6100 nm2 montre qu’environ 10 % de la surface est
occup´e par le domaine compl´ementaire ce qui est en accord avec d’autres ´etudes men´ees par STM [14].
En outre, il existe ´egalement sur cette surface des protub´erances qui repr´esentent tout de mˆeme 5 % de
la surface total mais qui ne contribuent pas au signal d’anisotropie. Le signal d’une surface id´eale doit
alors pr´esenter une anisotropie plus importante que celle obtenue ici car la pr´esence des protub´erances
entraˆıne une sous-estimation du signal d’anisotropie de 5 %. D’autre part, en tenant compte des 10 %
de la surface couverte par le domaine compl´ementaire, le signal d’anisotropie d’un surface id´eale peut
ˆetre extrapol´e `a partir du spectre de la figure 3.3 (b) mesur´e sur un surface fraˆıchement pr´epar´ee. On
obtient alors le spectre en vert sur la figure 3.3 (b) not´e (∆r
r )ideal et dont l’anisotropie maximale pic
`a pic atteint 5.5×10−3, ce qui n’a jamais pu ˆetre observ´e exp´erimentalement jusqu’`a pr´esent. Ainsi,
le signal d’anisotropie enregistr´e sur une surface de silicium (001) est une combinaison lin´eaire des
signaux provenant des deux domaines d’orientation diff´erente et peut s’´ecrire selon l’expression :
∆r
r
= A(
∆r
r
)ideal − (1 − A)(
∆r
r
)ideal (3.1)
o`u A repr´esente le pourcentage du domaine majoritaire sur la surface.
Pour valider cette hypoth`ese, j’ai mesur´e le spectre de RA d’une surface vicinale de Si(001) d´esorient´ee](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-35-320.jpg)
![16 CHAPITRE 3. LA SURFACE SI(001)-(2×1)
de 2◦ et fraˆıchement pr´epar´ee ; pr´esent´ee sur la figure 3.4 (a) (cercles). Le mode de pr´eparation de la
surface ´etant identique `a celui utilis´e pour la surface vicinale 4◦, le signal de RA a ´et´e augment´e de
5% pour tenir compte de la pr´esence des protub´erances. Le spectre simul´e, pr´esent´e sur la figure 3.4
(a), a ´et´e obtenu en consid´erant que 24% de la surface est occup´ee par le second domaine ; le signal
de RA de la surface de silicium vicinal 2◦ not´e (∆r
r )2◦ est ainsi reproduit en employant la formule :
(
∆r
r
)2◦ = 0.76(
∆r
r
)ideal − 0.24(
∆r
r
)ideal (3.2)
L’accord entre les deux courbes est tr`es satisfaisant pour des ´energies sup´erieures `a 2.5 eV, en revanche
`a basse ´energie l’accord est moins bon en raison de la pr´esence de l’´etat de surface qui est tr`es sensible
`a la largeur des terrasses. La technique RAS a permis d’estimer que le second domaine, minoritaire
pour l’orientation des dim`eres, recouvre 24% de la surface, ce qui est en accord avec une ´etude par
STM [14] o`u la proportion du seconde domaine est estim´ee `a 25%. Le mˆeme type de simulation a ´et´e
r´ealis´e pour la surface de silicium vicinal 4◦, d´et´erior´ee par de nombreux flashes, et les r´esultats sont
pr´esent´es sur la figure 3.4 (b). Pour tenir compte de la pr´esence de nombreuses protub´erances qui
repr´esentent 15% de la surface totale, le spectre exp´erimental a ´et´e augment´e de 15%. Dans ce cas,
l’anisotropie r´esultante provient uniquement de la diff´erence de r´eflectivit´e anisotrope entre les deux
domaines. On note que la courbe simul´ee (ligne verte) not´ee (∆r
r )4◦ reproduit parfaitement le spectre
exp´erimental (cercles bleu) ; le meilleur ajustement est obtenu en utilisant la formule :
(
∆r
r
)4◦ = 0.77(
∆r
r
)ideal − 0.23(
∆r
r
)ideal (3.3)
o`u 23% de la surface est occup´e par le second domaine minoritaire. On voit ici clairement que lorsque
la surface est r´eg´en´er´ee de nombreuses fois par des flashes successifs `a plus de 1000◦C, la qualit´e de la
surface se d´et´eriore rapidement et pr´esente un pourcentage non n´egligeable de dim`eres orient´es `a 90◦.
Des estimations similaires peuvent ˆetre obtenues dans le cas de surfaces nominales en utilisant comme
r´ef´erence, le spectre obtenu par Jaloviar et al. en appliquant des tensions de surface et o`u 92 % de la
surface est recouverte par une reconstruction 2×1 et 8 % par une reconstruction 1×2.
On se rend compte dans cette section que la technique RAS est un outil id´eal pour quantifier le ratio
des domaines qui coexistent sur la surface de Si(001)-(2×1) suppos´ee monodomaine.
3.3.3 Taux d’occupation des dim`eres.
Lorsque les mol´ecules qui sont adsorb´ees sur la surface de silicium ne pr´esentent pas de transitions
´electroniques dans le domaine U.V. visible, alors les spectres de r´eflectivit´e RAS ou SRDS, refl`etent la
modification de la r´eflectivit´e de la surface au cours de l’adsorption. Il est alors possible de montrer que
le signal optique est proportionnel au taux d’occupation des dim`eres de silicium, faisant de la RAS et
de la SRDS, des techniques particuli`erement adapt´ees pour ´etudier les cin´etiques d’adsorption. Les cas
de l’adsorption de mol´ecules de benz`ene et d’hydrog`ene atomique sont pr´esent´es pour illustrer cette
propri´et´e. En effet, dans ces deux syst`emes, les spectres RAS obtenus apr`es saturation de la surface
ne pr´esentent pas de structures provenant des adsorbats, ainsi le signal optique r´esulte uniquement
de la modification de la surface de silicium sous l’effet de l’adsorption. En ce qui concerne le syst`eme
benz`ene/Si(001)-2×1, de pr´ec´edentes ´etudes ont montr´e que les mol´ecules de benz`ene s’adsorbent un
dim`ere sur deux, laissant ainsi la moiti´e des dim`eres de la surface intacts, le taux d’occupation des
dim`eres est dans ce cas proche de 0.5 [15, 16, 17], ce syst`eme sera trait´e plus en d´etail dans la section 5.3.
En revanche lorsque la surface est expos´ee `a de l’hydrog`ene atomique `a 320◦C, chaque liaison pendante
du dim`ere se lie `a un atome d’hydrog`ene et l’occupation des dim`eres est alors pratiquement compl`ete,
ce syst`eme sera trait´e plus largement dans la section 4.1. La figure 3.5 (a) pr´esente les spectres de](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-36-320.jpg)
![3.3. PROPRI´ET´ES OPTIQUES 17
-1.5x10
-3
-1.0
-0.5
0.0
0.5
Re(∆r/r)
5.04.54.03.53.02.52.0
Energie de photon (eV)
(a)
1/2 MC hydrogène
saturé hydrogène
benzène
(b)
-0.8x10
-3
-0.7
-0.6
-0.5
-0.4
Signal RA @ 3.35 eV
2.01.51.00.50.0
Dose de benzène (L)
Fig. 3.5 — (a) : Spectre de RA enregistr´e sur une surface Si(001) vicinale 4◦ satur´ee en benz`ene, avec
0.5 MC d’hydrog`ene et satur´ee en hydrog`ene ; (b) : Cin´etique d’adsorption du benz`ene `a 3.35 eV.
RAS pour une surface satur´ee en benz`ene (violet), une surface partiellement recouverte d’hydrog`ene
atomique (orange) et une surface satur´ee en hydrog`ene atomique (rouge). On remarque que pour des
´energies de photon sup´erieures `a 3 eV, les spectres mesur´es apr`es saturation par du benz`ene et avec
0.5 MC (monocouche) d’hydrog`ene atomique sont tr`es semblables. Ils se composent d’une structure `a
4.4 eV similaire `a celle obtenue sur la surface propre au point critique E2, on distingue ´egalement sur les
deux spectres la pr´esence d’un ´epaulement vers 3.5 eV suivi d’un minimum vers 3.2 eV. La diff´erence
majeure entre les deux spectres se situe pour des ´energies plus faibles, dans une gamme d’´energie
faisant intervenir des ´etats ´electroniques purement de surface et donc tr`es sensibles aux changements
d’environnement local. Cette diff´erence peut alors r´esulter de la diff´erence qui existe entre une liaison
Si-C et une liaison Si-H. Lorsque la surface est `a pr´esent satur´ee par de l’hydrog`ene atomique, le profil
du spectre est fortement modifi´ee. On note plus particuli`erement la pr´esence d’une nouvelle structure
positive `a 3.5 eV, en retirant pour chaque spectre, le spectre correspondant `a une surface propre, il est
alors possible de mettre en ´evidence que l’amplitude du signal `a 3.5 eV est proportionnel `a l’occupation
des dim`eres de silicium. En effet, si l’on consid`ere que dans le cas de la surface satur´ee par l’hydrog`ene,
tous les dim`eres sont occup´es, alors dans le cas du benz`ene, on trouve un taux de recouvrement de
0.47±0.05 pour les dim`eres de silicium ce qui est en accord avec les pr´ec´edentes ´etudes.
Grˆace `a cette ´etude, il devient possible d’´etudier les cin´etiques d’adsorption sur la surface de silicium
car il est facile de suivre l’´evolution du signal RAS au cours de l’adsorption, comme le montre les
r´esultats pr´esent´es sur la figure 3.5 (b) o`u la cin´etique d’adsorption du benz`ene a ´et´e mesur´ee par
RAS pour une ´energie de photon de 3.4 eV. L’amplitude du signal `a 3.4 eV a ´et´e normalis´ee par le
signal enregistr´e sur une surface satur´ee en hydrog`ene en consid´erant un taux d’occupation des dim`eres
de 1. On remarque alors que comme pr´ec´edemment, le signal relatif au benz`ene atteint un taux de
couverture d’environ 0.5, validant ainsi l’´etude pr´ec´edente. D’autre part, on s’aper¸coit que le signal croit
de mani`ere exponentielle comme on peut l’observer dans le cas d’un processus de type Langmuirien o`u
les mol´ecules de benz`ene en arrivant sur la surface s’adsorbent directement ou d´esorbent de la surface.
Il est possible d’ajuster la courbe de cin´etique du benz`ene par une courbe exponentielle indiquant qu’il
s’agit bien ici d’un processus d’adsorption de type Langmuirien ; la courbe est correctement reproduite
en int´egrant l’´equation 3.4 :
dθ
dx
=
[D]
σ
√
2πmkT
S0e−Ea
kT (1 − θ) (3.4)
o`u θ est le taux de couverture, x est la dose en langmuir, [D] est un facteur correctif de pression, σ
est la densit´e de sites d’adsorption, S0 est le coefficient de collage initial et Ea est l’´energie d’activation.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-37-320.jpg)
![18 CHAPITRE 3. LA SURFACE SI(001)-(2×1)
1.0x10
-2
0.8
0.6
0.4
0.2
0.0
∆R/R
5.04.54.03.53.02.52.0
Energie (eV)
saturée en hydrogène
saturée en benzène
0.6x10
-2
0.5
0.4
0.3
0.2
0.1
0.0
∆R/R @2.76 eV
1.61.20.80.40.0
Dose de benzène (L)
0.07 0.330.25 0.42
Taux de dimËres occupÈs
(a) (b)
Fig. 3.6 — (a) : Spectres de SRDS enregistr´es sur une surface Si(001) vicinale 4◦ apr`es saturation par
de l’hydrog`ene atomique et du benz`ene. (b) : Cin´etique d’adsorption du benz`ene mesur´ee par SRDS
`a 2.76 eV.
La technique SDRS peut ´egalement ˆetre utilis´ee pour d´eterminer le taux de couverture de la surface de
silicium. La figure 3.6 (a) pr´esente les spectres SRDS mesur´es sur une surface de Si(001)-2×1 vicinale,
satur´ee par de l’hydrog`ene atomique `a 320◦C (courbe rouge) et satur´ee par du benz`ene (courbe bleu).
On remarque ici, que les deux spectres ont des formes tr`es similaires ; le spectre correspondant `a la
surface hydrog´en´ee sera ´etudi´e plus en d´etail par la suite. En revanche, l’intensit´e du signal SDRS pour
le benz`ene est pratiquement 2 fois moins intense que pour l’hydrog`ene. En consid´erant un taux de
couverture de 1 pour l’hydrog`ene, le taux de couverture des dim`eres par les mol´ecules de benz`ene peut
ˆetre de nouveau estim´ee `a 0.47, ce qui est en bon accord avec des mesures pr´ec´edentes. Comme dans le
cas de la RAS, il est possible de suivre l’´evolution du signal SRDS en fonction de la dose d’adsorbat.
Le signal enregistr´e `a 2.76 eV en fonction de la dose de benz`ene est pr´esent´e sur la figure 3.6, comme
dans le cas de la RAS, on voit que le signal est correctement reproduit par une courbe exponen-
tielle d´ecrite par l’´equation 3.4, indiquant de nouveau un processus d’adsorption de type Langmuirien.
Toutefois `a la fois en RAS et e SRDS, il est difficile d’extraire de ces courbes calcul´ees les param`etres
thermodynamiques d’adsorption en raison des difficult´es pour mesurer les pressions absolues lors de
l’enregistrement des spectres.
Ce chapitre permet de rendre compte des potentialit´es des spectroscopies optiques de surface quant
aux informations qui peuvent ˆetre obtenues sur la surface propre de silicium (001)-(2×1) et lors de
l’adsorption d’atomes ou de mol´ecules sur celle-ci. De mani`ere non exhaustive, ces techniques optiques,
tr`es faciles d’utilisation, permettent d’obtenir des informations pr´ecieuses sur la qualit´e globale de la
surface et la proportion de surface monodomaine ; elles peuvent s’av´erer utiles pour connaˆıtre le taux
d’occupation des dim`eres de silicium et sont de ce fait particuli`erement bien adapt´ees pour ´etudier les
cin´etiques d’adsorption. Ces r´esultats ainsi que d’autres concernant la contamination de la surface ont
r´ecemment ´et´e publi´e dans l’article [1] de l’annexe.
Dans la partie suivante, ces potentialit´es sont exploit´ees en compl´ement de calculs ab initio et de
mesures d’absorption X pour ´etudier l’adsorption d’atomes et de mol´ecules de complexit´e croissante
sur la surface Si(001)-(2 × 1).](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-38-320.jpg)
![Chapitre 4
Mol´ecules simples : hydrog`ene, oxyg`ene
4.1 Hydrog`ene atomique
4.1.1 Le syst`eme H/Si(001)-(2×1)
L’adsorption d’hydrog`ene atomique sur la surface Si(001)-(2×1) est ´etudi´ee depuis de nombreuses
ann´ees en raison de l’importance des liaisons hydrog`enes dans la chimie de cette surface [18]. Il est
`a pr´esent ´etabli que l’hydrog´enation de la surface `a diff´erentes temp´eratures donne lieu `a des recon-
structions pr´eservant ou non l’int´egrit´e des dim`eres de silicium. Ainsi, lorsque la surface est expos´ee
`a l’hydrog`ene atomique `a haute temp´erature, environ 300-350◦C, chaque liaison pendante se remplit
avec un atome d’hydrog`ene tout en pr´eservant le dim`ere. La surface pr´esente alors une reconstruction
ordonn´ee (2×1) identique `a la surface nue comme on peut le voir sur la figure 4.1(A). En revanche,
lorsque l’adsorption s’effectue `a temp´erature ambiante, la liaison du dim`ere est rompue et deux atomes
d’hydrog`ene se lient aux atomes de silicium de la surface. Celle-ci devient alors plus d´esordonn´ee et
pr´esente une reconstruction de type (1×1) comme indiqu´e sur la figure 4.1.B. Si l’adsorption s’effectue
`a une temp´erature interm´ediaire d’environ 125◦C, la surface pr´esente alors une reconstruction de type
(3×1), qui pr´esente une alternance de rang´ees monohydrides et dihydrides comme illustr´e sur la fig-
ure 4.1.C. Les surfaces de silicium hydrog´en´ees sont d’un grand int´erˆet car en saturant les liaisons
pendantes, l’hydrog`ene passive la surface la rendant moins r´eactive et plus adapt´ee `a la croissance
de compos´es organiques plus complexes. Dans les cas des surfaces monohydrides et de reconstruction
(3×1), on voit qu’il est possible de pr´eserver la nanostructuration de la surface faisant de ces sur-
faces de bonnes candidates pour d´evelopper l’assemblage de structures supramol´eculaires ayant des
A : Si(001)-(2x1)-H B : Si(001)-(1x1)-H C : Si(001)-(3x1)-H
Fig. 4.1 — Repr´esentations sch´ematiques des surfaces monohydride (A), dihydride (B) et reconstruite
(3×1) (C).
19](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-39-320.jpg)
![20 CHAPITRE 4. MOL´ECULES SIMPLES : HYDROG`ENE, OXYG`ENE
300
250
200
150
100
50
0
Temps (s)
5.04.54.03.53.02.52.01.5
Energie (eV)
300
250
200
150
100
50
0
Temps (s)
5.04.54.03.53.02.52.01.5
Energie (eV)
12x10
-3
10
8
6
4
2
0
∆R/R
5.04.54.03.53.02.52.01.5
Energie (eV)
après 100 s
après 300 s
12x10
-3
10
8
6
4
2
0
∆R/R
5.04.54.03.53.02.52.01.5
Energie (eV)
après 300 s
320°C 20°C(A) (B)
(C) (D)
Fig. 4.2 — (A) Spectres de SDRS en fonction du temps pour l’hydrog´enation `a 320◦C et (B) l’hy-
drog´enation `a temp´erature ambiante ; (C) Spectre de SRDS apr`es 300 s `a 320◦C ; (D) Spectres de
SRDS apr`es 100 s et 300 s `a temp´erature ambiante.
propri´et´es optiques et ´electroniques originales. Dans cette partie, on s’int´eresse plus particuli`erement
aux propri´et´es optiques des surfaces hydrog´en´ees et on verra comment le clivage des dim`eres lors de
l’adsorption `a temp´erature ambiante peut ˆetre mis en ´evidence.
4.1.2 Propri´et´es optiques
La technique SRDS permet d’enregistrer les spectres en temps r´eel lors de l’adsorption de l’hy-
drog`ene. L’hydrog´enation d’une surface Si(001)-(2×1) vicinale, a ´et´e suivie `a 320◦C et `a temp´erature
ambiante sous une pression d’hydrog`ene de 10−6 T ; les spectres correspondants sont repr´esent´es sur la
figure 4.2. Les figures (A) et (B) pr´esentent l’ensemble des spectres enregistr´es en fonction du temps,
l’amplitude ∆R
R est repr´esent´ee par des couleurs allant de l’indigo au rouge pour des amplitudes crois-
santes. Les deux figures (A) et (B) sont clairement diff´erentes : `a 320◦C, une seule structure principale
situ´ee `a 2.8 eV apparait apr`es une centaine de secondes et sature tr`es rapidement ; `a temp´erature
ambiante, une premi`ere structure apparaˆıt `a 3 eV environ et par la suite une deuxi`eme structure se
d´eveloppe `a 4 eV. Pour plus de pr´ecision, les figures (C) et (D) repr´esentent respectivement les spectres
obtenus `a saturation pour l’hydrog´enation `a 320◦C, et les spectres obtenus apr`es 100 s et apr`es sat-
uration pour l’hydrog´enation `a temp´erature ambiante. Le spectre obtenu sur la surface monohydride
(C) pr´esente une structure principale autour de 2.8 eV ainsi qu’un ´epaulement autour de 3.8 eV. En
revanche, `a temp´erature ambiante, le spectre `a saturation pr´esente deux structures bien marqu´ees `a
3 eV et 4 eV. En raison de la diff´erence de temp´erature, la structure principale `a 2.8 eV qui est ob-
serv´ee `a 320◦C est d´ecal´ee vers de plus hautes ´energies `a temp´erature ambiante. Le creux qui apparaˆıt
`a 3.5 eV correspond aux points critiques E1, E0, qui sont sensibles aux tensions sur la surface [11].
L’´evolution du signal SRDS en fonction du temps montre clairement qu’apr`es avoir expos´e la surface
pendant une centaine de seconde `a l’hydrog`ene atomique `a temp´erature ambiante, le spectre SRDS
ressemble au spectre obtenu sur la surface monohydride. En effet, on remarque que l’hydrog´enation](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-40-320.jpg)
![4.1. HYDROG`ENE ATOMIQUE 21
`a temp´erature ambiante s’effectue en deux temps : dans un premier temps les atomes d’hydrog`ene
viennent saturer les liaisons pendantes puis dans un deuxi`eme temps, le dim`ere se casse et chaque
atome de silicium se lie avec deux atomes d’hydrog`ene formant ainsi une surface dihydrog´en´ee. Ce
processus avait d´ej`a ´et´e mis en ´evidence par des ´etudes STM [18] mais aucune observation directe du
passage monohydride `a dihydride `a temp´erature ambiante n’avait pu ˆetre faite en raison de la difficult´e
`a ´etudier l’hydrog´enation en temps r´eel.
4.1.3 Evidence de la cassure des dim`eres de silicium `a temp´erature ambiante
Comme on vient de le voir, les spectres optiques SRDS sont tr`es diff´erents sur une surface mono-
hydride o`u les dim`eres sont pr´eserv´es et sur une surface dihydride o`u les dim`eres sont cliv´es. L’´etude
de l’hydrog´enation de la surface de silicium apparaˆıt comme ´etant un syst`eme mod`ele pour mettre en
´evidence la cassure des dim`eres de silicium au cours des processus d’adsorption. Cette information est
particuli`erement importante pour comprendre les m´ecanismes qui r´egissent l’adsorption des mol´ecules
sur les surfaces. Or, les techniques de surface habituelles telles que la diffraction d’´electrons lents ou
la microscopie en champ proche peinent `a renseigner sur les propri´et´es structurales de l’interface pour
des taux de recouvrement important car l’acc`es `a l’interface est compliqu´e par la pr´esence de la couche
d’adsorbat. En revanche, les techniques utilisant des photons peuvent ˆetre utilis´ees pour sonder l’inter-
face au travers de la couche adsorb´ee. Ainsi, la spectroscopie infrarouge est couramment utilis´ee pour
´etudier les liaisons mol´ecules-silicium ainsi que les modes de vibration des mol´ecules mais ne peut pas
donner d’information quant aux liaisons Si-Si elles-mˆemes [19]. Les techniques utilisant le rayonnement
synchrotron, telles que la diffraction et les spectroscopies de photo´electrons [20, 21], ont montr´e leur
puissance pour caract´eriser les liaisons entre substrat et adsorbat, comme il sera pr´esent´e plus loin
dans ce manuscrit. Toutefois, ces techniques n´ecessitent l’utilisation du rayonnement synchrotron et
ne peuvent pas ˆetre utilis´ees de mani`ere quotidienne. Les r´esultats expos´es dans la suite d´emontrent
que la technique SRDS permet de d´eterminer de mani`ere tr`es claire si il y a pr´eservation ou non des
dim`eres de silicium lors de l’adsorption de mol´ecules organiques.
Pour ce faire, nous avons compar´e les r´esultats obtenus sur les deux syst`emes mod`eles qui sont la
surface de silicium monohydride Si(001)-(2×1) : H, o`u les dim`eres sont pr´eserv´es ; et la surface de
silicium dihydride Si(001)-(1×1) : H, o`u les dim`eres sont bris´es au cours de l’adsorption. Les spectres
(a) et (b) de la figure 4.3 ont ´et´e enregistr´es respectivement sur une surface monohydride et sur une
surface dihydride, et sont d´efinis tels que :
∆R
R
=
RSi − RSi:H
RSi
(4.1)
avec RSi et RSi:H repr´esentant respectivement les r´eflectances de la surface propre et de la surface
hydrog´en´ee. La r´eflectance RSi de la surface propre Si(001)-(2×1) comporte une contribution provenant
du volume et une contribution provenant de la surface, caract´eris´ee par la reconstruction (2×1) et la
pr´esence d’´etats de surface [22]. Comme l’indiquent des calculs DFT r´ealis´es sur cette surface [9], les
transitions ´electroniques peuvent avoir diff´erentes origines ; il peut s’agir de transitions entre ´etats de
volume au voisinage de la surface, qui, du fait de la pr´esence de la brisure de sym´etrie provoqu´ee par
la surface, peuvent ˆetre diff´erents des ´etats du volume, et peuvent participer ainsi `a la contribution de
surface de la r´eflectance. Ces transitions de volume modifi´ees par la surface ont lieu pour des ´energies
correspondant aux points critiques et sont sensibles `a des modifications cons´equentes de la surface.
Il peut s’agir ´egalement de transitions impliquant directement des ´etats de surface, ces derniers sont
particuli`erement sensibles aux modifications de la surface provoqu´ees par l’adsorption de mol´ecules par
exemple. Ainsi, quand la surface de silicium reconstruite (2 × 1) est expos´ee `a l’hydrog`ene atomique,
ces diff´erentes transitions vont ˆetre modifi´ees, voire supprim´ees, provoquant des modifications dans](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-41-320.jpg)
![22 CHAPITRE 4. MOL´ECULES SIMPLES : HYDROG`ENE, OXYG`ENE
∆R/R
5.04.03.02.0
Energie (eV)
5.04.03.02.0
Energie (eV)
0
0
0
0
4.10-3
4.10-3 4.10-3
1.10-2
(a)
(b)
(c)
(d)
Fig. 4.3 — (a) Spectre de SDRS apr`es saturation de la surface par de l’hydrog`ene atomique `a 320 ◦C ; (b)
spectre de SDRS apr`es saturation de la surface par de l’hydrog`ene atomique `a temp´erature ambiante,
(c) Calcul DFT correspondant au spectre (b) d’apr`es [9] ; (d) Contribution au signal SRDS de la
cassure des dim`eres.
le spectre SRDS. Lorsque l’hydrog`ene est adsorb´e `a haute temp´erature, le spectre correspondant
(a) de la figure 4.3 pr´esente un pic large vers 2.9 eV avec un ´epaulement. La surface de silicium
r´esultante est pratiquement monohydrog´en´ee, c’est `a dire que chaque liaison pendante est occup´ee
par un atome d’hydrog`ene tout en pr´eservant l’int´egrit´e du dim`ere. Ainsi, dans le spectre (a) seules
les transitions impliquant les ´etats ´electroniques des liaisons pendantes ont ´et´e modifi´ees et ce spectre
peut alors ˆetre consid´er´e comme caract´eristique de l’adsorption sur les liaisons pendantes. L’absence de
structures aux ´energies correspondant aux points critiques E0, E1 `a 3.45 eV et E2 `a 4.35 eV indique
qu’aucune transition de volume modifi´ee par la surface n’a ´et´e impliqu´ee lors de cette adsorption
confirmant la seule participation des ´etats de surface dans ce processus. Lorsque l’hydrog`ene est
adsorb´e `a temp´erature ambiante, le spectre correspondant (b) de la figure 4.3 pr´esente deux pics `a
3 eV et 4 eV et un creux `a 3.5 eV. La surface de silicium r´esultante est `a pr´esent dihydrog´en´ee, c’est `a
dire que chaque atome de silicium de la surface est li´e `a deux atomes d’hydrog`ene entraˆınant le clivage
des dim`eres de silicium et ainsi la disparition de la reconstruction (2×1). Comme la r´eflectance de la
surface dihydride RSi−1×1:H est tr`es proche de celle du volume, ce spectre peut ˆetre consid´er´e comme
la r´eponse optique intrins`eque d’une surface nue de Si(001)-(2×1). Ce spectre est en bon accord avec
les r´esultats de calculs DFT effectu´es sur cette surface [9] repr´esent´e sur le spectre (c). Afin d’isoler
la contribution du spectre SDR provenant du clivage des dim`eres, le spectre correspondant `a une
adsorption initiale sur les liaisons pendantes, similaire au spectre (a), a ´et´e soustrait du spectre (b)
mesur´e sur la surface dihydride. Cette diff´erence correspond au spectre (d) de la figure 4.3 et se compose
de deux pics `a 3 eV et 4.5 eV et d’un creux `a 3.5 eV. Ainsi ce spectre, qui pr´esente une structuration
`a l’´energie des points critiques, peut ˆetre consid´er´e comme l’empreinte optique du clivage des dim`eres
de silicium r´esultant de la suppression des transitions de surface relatives aux ´etats ´electroniques des
dim`eres et de la suppression des ´etats de volume provoqu´es par la disparition de la reconstruction
(2×1).
Cette approche ph´enom´enologique fournit une m´ethode in´edite pour pouvoir distinguer l’adsorption
sur les liaisons pendantes des dim`eres de silicium, de l’adsorption provoquant le clivage ou le pontage
des dim`eres de silicium. Ces r´esultats ont ´et´e test´es sur d’autres mol´ecules et sont d´etaill´es dans l’article
[2] de l’annexe.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-42-320.jpg)
![4.2. OXYG`ENE 23
4.2 Oxyg`ene
4.2.1 Le syst`eme O2/Si(001)-(2×1)
L’´etude de l’oxydation du silicium a fait l’objet de tr`es nombreux travaux en raison de l’int´erˆet tech-
nologique du Si(001), dans les syst`emes micro´electroniques actuels. En particulier, le d´eveloppement
grandissant des nouveaux transistors organiques impose que la couche isolante de SiO2 atteigne seule-
ment quelques nanom`etres. Il devient alors crucial de comprendre comment se forme l’interface Si/SiO2
pour de faibles taux d’exposition `a l’oxyg`ene afin d’en contrˆoler la formation. La compr´ehension des
processus impliqu´es dans l’oxydation initiale du Si(001) est encore actuellement incompl`ete et en
particulier, les sites d’adsorption, les modifications structurales de la surface ou encore les chemins
r´eactionnels, font l’objet de d´ebats et de controverses [23].
Ces derni`eres ann´ees, les premiers stades de l’oxydation `a temp´erature ambiante de la surface Si(001)-
(2×1) ont ´et´e ´etudi´es par des techniques exp´erimentales nombreuses et vari´ees. Ainsi, une ´etude
combinant microscopie ´electronique en r´eflexion, spectroscopie Auger et photo´emission, a montr´e que
l’oxydation de la surface de Si(001) a lieu couche par couche et que la premi`ere couche de silicium
est oxyd´ee par de l’oxyg`ene mol´eculaire et cela sans barri`ere ´energ´etique [24]. Dans une autre ´etude,
le profil d’incorporation de l’oxyg`ene a pu ˆetre suivi d`es les tous premiers stades de l’oxydation par
spectroscopie de r´etro diffusion Rutherford `a haute r´esolution (HRBS) [25]. Ainsi, dans cette ´etude,
il apparaˆıt que l’oxyg`ene s’adsorbe principalement sur la premi`ere couche du silicium, la saturation
´etant atteinte pour environ 1.5 MC. D’autres ´etudes men´ees par diffraction de photo´electrons [26],
photo´emission [27] et photo´emission r´esolue en temps [28] ont ´et´e utilis´ees pour caract´eriser l’´etat
d’oxydation des atomes de silicium impliqu´es dans les liaisons avec les atomes d’oxyg`ene, `a la fois
`a la surface et `a l’interface oxyde/silicium, elles ont pu d´emontrer l’existence d’une liaison du type
Si-O-Si `a l’interface. D’autre part, une ´etude in situ men´ee par STM/STS `a temp´erature ambiante
avec de l’oxyg`ene mol´eculaire sugg`ere que les atomes d’oxyg`ene s’adsorbent initialement sur les li-
aisons arri`eres des dim`eres de silicium. Cette ´etude montre en outre que pour une exposition de 4.5 L,
la reconstruction (2×1) de la surface est toujours pr´esente en raison de l’existence de domaines de
la surface o`u les dim`eres sont inoxyd´es [29]. Une ´etude plus r´ecente r´ealis´ee en utilisant de l’ozone
plutˆot que de l’oxyg`ene mol´eculaire, montre que l’adsorption initiale a lieu de mani`ere pr´ef´erentielle
sur le dim`ere de silicium en position pont´ee et sur les liaisons arri`ere des dim`eres [30]. Ces r´esultats
sont confirm´es par des calculs ab initio d’´energie d’adsorption r´ealis´es par diff´erents auteurs utilisant
des m´ethodes DFT-LDA ou des m´ethodes de chimie quantique [31, 32, 33, 34, 35]. Une autre ´etude
th´eorique men´ee par Ciacchi et al. sur la base de calculs de dynamique mol´eculaire a ´et´e appliqu´ee `a
la croissance de l’oxyde natif du Si(001) et montre qu’apr`es la dissociation spontan´ee des mol´ecules
d’O2, les atomes d’oxyg`ene peuvent ˆetre captur´es sur les dim`eres ou sur les liaisons arri`eres ; et en
particulier, deux atomes d’oxyg`ene peuvent occuper deux dim`eres adjacents de la surface [23]. Ainsi,
nombres d’´etudes tendent `a montrer que l’oxyg`ene mol´eculaire se dissocie en arrivant sur la surface
de silicium ; les atomes d’oxyg`ene forment alors des liaisons Si-O-Si sur les dim`eres de silicium et leurs
liaisons arri`ere. Toutefois, des incertitudes demeurent sur les m´ecanismes qui r´egissent l’incorporation
de l’oxyg`ene sur ces deux types de site et leur cin´etique d’adsorption.
Depuis une dizaine d’ann´ee d´ej`a, des ´etudes ont port´e sur la modification des propri´et´es optiques de
la surface de Si(001)-(2×1) lors de son exposition `a l’oxyg`ene mol´eculaire [36]. Toutefois, ces ´etudes
visaient `a mieux comprendre la r´eponse optique de la surface de silicium par RAS plutˆot que les
m´ecanismes d’adsorption de l’oxyg`ene. Les r´esultats pr´esent´es dans la suite d´emontrent qu’en combi-
nant les spectres RA exp´erimentaux, pour diff´erents taux d’exposition `a l’oxyg`ene, avec des calculs
ab initio r´ealis´es en consid´erant plusieurs sites d’adsorption, il est possible de mieux comprendre les
processus r´egissant l’oxydation initiale de la surface Si(001)-(2×1).](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-43-320.jpg)
![24 CHAPITRE 4. MOL´ECULES SIMPLES : HYDROG`ENE, OXYG`ENE
4.2.2 M´ecanismes microscopiques lors de l’oxydation
C1 C2 1D 1E
signalRA(%)
Energie des photons
propre
Fig. 4.4 — Haut : sites d’adorption d’atomes d’oxyg`ene incorpor´es `a la surface du Si(001) ; Bas :
spectres de RA correspondant aux diff´erents sites d’adsorption de l’oxyg`ene
Les calculs ab initio pr´esent´es ici ont ´et´e obtenus grˆace `a des collaborations avec les ´equipes de G.
Onida de Milan, F. Bechstedt de Jena et R. Del Sole de Rome. Les g´eom´etries relax´ees `a l’´equilibre
qui sont repr´esent´ees sur la figure 4.4, ont ´et´e calcul´ees par une m´ethode DFT standard en min-
imisant l’´energie totale dans une cellule (2×2). Les ´etats excit´es ont ´et´e obtenus dans le cadre de
l’approximation ind´ependante des quasi-particules, en utilisant les valeurs propres et vecteurs propres
de Kohn-Sham comme point de d´epart et en n´egligeant les effets excitoniques. Plus de d´etails sur les
m´ethodes num´eriques utilis´ees peuvent ˆetre obtenus dans les articles suivants [5, 37].
Sur les quatre structures relax´ees de la figure 4.4, on remarque que dans la structure not´ee 1D, les
atomes d’oxyg`ene sont incorpor´es sur tous les dim`eres ; dans les structures 1E et C2 qui sont tr`es
semblables, les liaisons arri`eres seulement sont occup´ees, et dans la structure C2, `a la fois les dim`eres
et les liaisons arri`ere peuvent ˆetre li´es `a un atome d’oxyg`ene. Les spectres de RA correspondants `a ces
quatre structures pour un taux de couverture de 0.5 MC, ainsi que le spectre calcul´e pour la surface
propre sont donn´es dans la figure 4.4. Bien que les quatre spectres aient ´et´e obtenus pour un mˆeme
taux de couverture de 0.5 MC, il est ´etonnant de remarquer de si grandes diff´erences entre eux. En
effet, lorsque l’oxyg`ene est incorpor´e sur les liaisons arri`ere comme dans les structures 1E et C2, alors
l’anisotropie optique est pratiquement supprim´ee et les spectres correspondant sont pratiquement
plats et sans structures notables. En revanche, lorsque l’oxyg`ene est adsorb´e sur le dim`ere comme
dans la configuration 1D, le spectre RA correspondant pr´esente des structures qui sont beaucoup plus
marqu´ees. Pour la structure mixte C1, o`u l’oxyg`ene s’adsorbe `a la fois sur le dim`ere et sur la liaison
arri`ere, alors le spectre est pratiquement inchang´e. A partir des spectres calcul´es pour les 4 struc-
tures relax´ees et du spectre de la surface propre, nos coll`egues ont essay´e de reproduire les spectres
exp´erimentaux mesur´es pour diff´erents taux d’exposition `a l’oxyg`ene. Les r´esultats de ces simulations
et les spectres exp´erimentaux sont pr´esent´es dans la figure 4.5.
Les spectres exp´erimentaux de la figure 4.5 ont ´et´e mesur´es sur une surface nominale, pr´epar´ee en
utilisant la proc´edure d’´electromigration expos´ee dans la partie 3.2.1 ; sur cette surface, environ 80 %](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-44-320.jpg)
![4.2. OXYG`ENE 25
Energie des photons
expérience
théorie
propre
signalRA(%)signalRA(%)
signalRA(%)signalRA(%)
Energie des photons
expérience
théorie
Fig. 4.5 — Spectres de RA exp´erimentaux et calcul´es pour diff´erents taux d’exposition `a l’oxyg`ene
des dim`eres sont orient´es dans la mˆeme direction. L’oxyg`ene mol´eculaire a ´et´e dos´e sur la surface
grˆace une vanne micro fuite et sa puret´e a ´et´e contrˆol´ee par spectrom´etrie de masse. Etant ´etabli que
les calculs th´eoriques de spectres optiques surestiment les intensit´es, tous les spectres calcul´es ont ´et´e
r´eduit d’un facteur 4.8 pour reproduire le plus fid`element possible les r´esultats exp´erimentaux [9, 38].
Sur la figure 4.5 sont expos´es les spectres exp´erimentaux et calcul´es pour la surface propre, et apr`es
des expositions successives d’oxyg`ene mol´eculaire de 1.3 L, 2.7 L et 4.7 L. On s’aper¸coit que dans le
cas de la surface propre, le spectre calcul´e est en tr`es bon accord avec le spectre exp´erimental, ce qui
valide la m´ethode de calcul utilis´ee. Pour les diff´erents taux d’exposition `a l’oxyg`ene, l’accord entre
spectres calcul´es et spectres exp´erimentaux est ´egalement tr`es bon ; toutefois aucunes des configura-
tions repr´esent´ees sur la figure 4.4 ne permet `a elle seule de reproduire les spectres exp´erimentaux. En
effet, comme les ´energies des structures propos´ees sont tr`es proches, il est tr`es probable que l’oxyg`ene
s’incorpore initialement dans diff´erents sites. Dans ce cas, il est plus r´ealiste de consid´erer que la
surface de silicium se compose de zones inoxyd´ees et de zones o`u l’oxyg`ene est incorpor´e selon les
structures repr´esent´ees sur la figure 4.4. C’est pourquoi il a fallu m´elanger les spectres des diff´erentes
structures et de la surface propre pour pouvoir obtenir un accord satisfaisant. Comme les spectres
calcul´es pour les structures C2 et 1E, o`u l’adsorption se fait sur les liaisons arri`ere, sont tr`es similaires,
c’est la structure 1E qui a l’´energie la plus faible qui a ´et´e privil´egi´ee. Le choix de l’autre structure
C2 donne des r´esultats qui sont tr`es semblables. Ainsi, pour reproduire les spectres exp´erimentaux
correspondant aux diff´erents taux de couverture, on a consid´er´e une combinaison lin´eaire des spectres
calcul´es pour les structures 1D 1E et C1, ainsi que le spectre calcul´e sur la surface propre. En con-
sid´erant que chacune des contributions recouvrent respectivement les fractions de surface xD, xE, xC
et (1 − xD − xE − xC) ; le spectre de RA s’´ecrit dans ces conditions de la mani`ere suivante :
∆r
r
= xD ×
∆r
r (1D)
+ xE ×
∆r
r (1E)
+ xC ×
∆r
r (C1)
+ (1 − xD − xE − xC) ×
∆r
r (clean)
(4.2)
avec les coefficients xD, xE et xC d´etermin´es grˆace `a un ajustement des courbes exp´erimentales des
diff´erents taux de d’exposition par la m´ethode des moindre carr´es.
On voit sur l’ensemble des spectres pr´esent´es sur la figure 4.5 que l’accord entre spectres calcul´es et
spectres exp´erimentaux est tr`es satisfaisant quelque soit le taux d’exposition `a l’oxyg`ene. La premi`ere
information d´eterminante d´eduite de l’ajustement des spectres exp´erimentaux est que le coefficient xC](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-45-320.jpg)
![26 CHAPITRE 4. MOL´ECULES SIMPLES : HYDROG`ENE, OXYG`ENE
Exposition
Tauxdecouverture
Coéfficients
propre
couverture
Fig. 4.6 — Evolution des coefficients utilis´es pour reproduire les spectres exp´erimentaux de la figure
4.5 en fonction de l’exposition et taux de couverture correspondant
reste ´egal `a z´ero quelque soit le taux d’exposition, cela indique de fa¸con non ´equivoque que l’oxyg`ene
ne peut pas s’incorporer `a la fois sur les dim`eres et les liaisons arri`eres, comme c’est le cas dans la
configuration C1. Ainsi, les calculs montrent que pour des faibles taux d’exposition, il existe des zones
o`u l’oxyg`ene s’adsorbe sur les dim`eres et d’autres zones o`u l’oxyg`ene s’adsorbe plutˆot sur les liaisons
arri`ere.
Pour essayer de mieux comprendre la cin´etique d’adsorption de l’oxyg`ene mol´eculaire, l’´evolution des
coefficients xD, xE, (1 − xD − xE) correspondant `a la fraction de surface propre, ainsi que le taux
de couverture donn´e par 0.5×(xD + xE) MC, ont ´et´e report´es sur la figure 4.6 en fonction du taux
d’exposition. On remarque ici une ´evolution r´eguli`ere tout `a fait remarquable des coefficients xD et xE
pour des expositions croissantes `a l’oxyg`ene, indiquant une augmentation progressive de la fraction
de la surface couverte par les structures 1D et 1E. D’apr`es le graphe de la figure 4.6, on peut estimer
qu’un taux de couverture de 0.47 MC est atteint pour une exposition de 4.7 L, ce qui est en bon accord
avec des r´esultats obtenus par d’autres techniques [24, 25]. De plus, il est possible de reproduire de
mani`ere tr`es satisfaisante l’´evolution des coefficients xD et xE, et donc ´egalement de la fraction de
surface propre, par des courbes de type exponentiel (repr´esent´es en trait plein sur la figure 4.6) qui
caract´erisent un comportement Langmuirien lors de l’incorporation de l’oxyg`ene. Ainsi, un m´ecanisme
Langmuirien d’adsorption peut ˆetre propos´e o`u les mol´ecules d’O2 arrivant sur une zone non oxyd´ee
de la surface de silicium, se dissocient et s’adsorbent soit dans une configuration de type 1E ou 1D
avec des probabilit´es d’incorporation diff´erentes pE et pD respectivement, telles que pD + pE = 1. En
suivant ces hypoth`eses, les coefficients xD et xE doivent respecter les ´equations diff´erentielles coupl´ees
suivantes :
dxD
dt
= pD(1 − xD − xE) (4.3)
dxE
dt
= pE(1 − xD − xE) (4.4)
o`u t correspond au temps d’exposition `a l’oxyg`ene.
La solution de ces ´equations coupl´ees est donn´e par :
xD,E = pD,E(1 − e−t/τ
) = pD,E(1 − e−δ/δ0
) (4.5)
o`u δ correspond `a l’exposition en Langmuir.
Les ´evolutions des coefficients xD et xE sont tr`es bien reproduites par le mod`ele propos´e, comme on
peut le voir sur la figure 4.6 (trait plein) en utilisant les param`etres suivants : pD=0.42 et pE = 0.58 et](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-46-320.jpg)
![4.2. OXYG`ENE 27
δ0=2.2 L. On s’aper¸coit que mˆeme si l’ordre de grandeur des probabilit´es d’adsorption dans les deux
structures 1D et 1E sont similaires, c’est l’adsorption dans la configuration 1E qui est avantag´ee.
La comparaison des r´esultats exp´erimentaux obtenus par la technique RAS coupl´ee `a des calculs
th´eoriques r´ealis´es en consid´erant plusieurs mod`eles d’incorporation de l’oxyg`ene nous a permis de
d´evelopper une nouvelle vision des premiers stades d’adsorption de l’oxyg`ene mol´eculaire sur la surface
de silicium Si(001)-(2×1). Deux m´ecanismes d’incorporation de l’oxyg`ene `a la surface du silicium ont
pu ˆetre mis en ´evidence : d’une part les atomes d’oxyg`ene peuvent s’adsorber sur les dim`eres de silicium,
d’autre part, ils s’adsorbent sur les liaisons arri`ere des dim`eres. L’analyse des donn´ees a montr´e que
la cin´etique des ces deux m´ecanismes suit un processus de type Langmuirien o`u les atomes d’oxyg`ene
arrivant sur une aire de la surface non oxyd´ee vont venir s’incorporer soit dans la configuration 1D soit
dans la configuration 1E, cette derni`ere ´etant l´eg`erement plus favorable d’apr`es le mod`ele utilis´e. Cette
m´ethode “tout optique” pour obtenir des informations structurales et cin´etiques lors de l’adsorption de
mol´ecules sur une surface, ouvre la voie `a l’´etude des m´ecanismes d’adsorption sur des types de surfaces
tels que les oxydes difficiles `a ´etudier par des m´ethodes conventionnelles de surface. Ces r´esultats sont
d´etaill´es dans l’article [3] de l’annexe.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-47-320.jpg)

![Chapitre 5
Mol´ecules organiques carbon´ees
5.1 Ac´etyl`ene
5.1.1 Le syst`eme C2H2/Si(001)-(2×1)
L’´etude de l’adsorption d’ac´etyl`ene sur Si(001)-(2×1), mol´ecule carbon´ee la plus simple poss´edant
une triple liaison C≡C, a d´ebut´e depuis la fin des ann´ees 80 en raison de son int´erˆet technologique
dans la fabrication de composants ´electroniques `a base de carbure de silicium. Dans une ´etude pr´ecoce,
Nishijima et al. [39], d´emontrent que l’ac´etyl`ene s’adsorbe sur le dim`ere de silicium sans dissociation
dans une configuration d´enomm´ee dimerized (figure 5.1. (a)) en utilisant la technique de spectroscopie
de perte d’´energie d’´electrons (HREELS) et la diffraction d’´electrons lents (LEED). Dans cette ´etude,
les liaisons pendantes sont satur´ees et le dim`ere de silicium reste intact lors de l’adsorption. Quelques
ann´ees plus tard, une ´etude regroupant la spectroscopie Auger, la d´esorption thermique (TPD) et
la technique HREELS [40, 41], montre que le dim`ere est cass´e lors de l’adsorption de l’ac´etyl`ene qui
pr´esente alors une configuration du type broken−dimer (figure 5.1. (b)) laissant les liaisons pendantes
vides. En ce qui concerne les ´etudes th´eoriques, la situation n’est pas tr`es claire. Des ´etudes anciennes
bas´ees sur des calculs classiques de couches ou utilisant les fonctionnelles de densit´e [42, 43] indiquent
que les configurations les plus stables sont respectivement celles o`u le dim`ere est cass´e et celle o`u
le dim`ere reste intact. Dans une autre ´etude de simulation d’images de microscopie `a effet tunnel
par Hofer et al. [44], la configuration dimerized est trouv´ee la plus stable. Outre ces deux modes
d’adsorption dimerized et broken − dimer, trois autres configurations d’adsorption ont ´egalement ´et´e
envisag´ees : le mod`ele end − bridge (figure 5.1. (c)), le mod`ele p − bridge (figure 5.1. (d)) et le mod`ele
r − bridge (figure 5.1. (e)). Dans le mod`ele end − bridge, deux mol´ecules d’ac´etyl`ene pontent deux
dim`eres adjacents, les atomes de carbones pr´esentent une hybridation du type sp2. Ainsi la double
liaison C-C est pr´eserv´ee et le dim`ere reste intact [45]. Les deux derniers mod`eles, propos´e par Xu et
al. [46] proposent des configurations o`u les atomes de carbones pr´esentent une hybridation de type
sp3 : la liaison C-C s’allonge de 0.2 ˚A en se transformant en simple liaison et la mol´ecule d’ac´etyl`ene
s’adsorbe sur deux dim`eres (tetra − σbonds). L’axe C-C de la mol´ecule peut alors ˆetre parall`ele aux
dim`eres (configuration p−bridge figure 5.1. (d)) ou perpendiculaire au dim`ere (configuration r−bridge
figure 5.1. (e)). Des ´etudes exp´erimentales ont montr´e que plusieurs de ces configurations pouvaient
co-exister sur la surface de Si(001)-(2×1).
Ainsi, une ´etude par diffraction de photo´electrons [47] met en ´evidence la pr´esence simultan´ee des
configurations dimerized et tetra − σbonds. A partir de la comparaison de mesures exp´erimentales
HREELS et de spectres calcul´es, une ´etude ult´erieure montre qu’`a bas taux de couverture la configura-
tion dimerized est plus stable alors qu’`a saturation c’est la configuration end − bridge qui pr´edomine
[48], corroborant les r´esultats obtenus par calculs th´eoriques de fonctionnelle de densit´e [49]. La coexis-
29](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-49-320.jpg)
![30 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
(a) dimerized
(b) broken-dimer
(c) end-bridge
(d) p-bridge
(e) p-bridge
[110]
[110]
y
x
z
Fig. 5.1 — Modes d’adsorption de l’ac´etyl`ene sur Si(001)-(2×1)
tence des configurations dimerized et end−bridge est aussi mise en ´evidence par des ´etudes th´eoriques
de type DFT-LDA [50, 51]. En revanche, des ´etudes men´ees par photo´emission sur les niveaux de cœur
n’ont pas pu mettre en ´evidence des configurations d’adsorption diff´erentes [52]. En effet, les configu-
rations dimerized et end − bridge sont chimiquement tr`es semblables et ne peuvent pas ˆetre r´esolues
par cette technique. Une pr´ec´edente ´etude par NEXAFS [53, 54] favorise une adsorption selon les
configurations dimerized et end − bridge, mais la d´etermination de la g´eom´etrie adopt´ee est limit´ee
par l’utilisation de surfaces pr´esentant les deux orientations de dim`eres. En effet, la d´etermination
de la configuration d’adsorption est compliqu´ee par la pr´esence de deux domaines tourn´es de 90◦ sur
la surface de silicium (001) reconstruite (2×1). Il devient alors difficile de discriminer deux configu-
rations similaires telles que dimerized et end − bridge ou p − bridge et r − bridge. C’est pourquoi
l’utilisation de surfaces monodomaines est d’un int´erˆet crucial pour d´eterminer la configuration d’ad-
sorption de l’ac´etyl`ene. Ainsi, en d´epit des nombreuses ´etudes qui ont ´et´e men´ees pour d´eterminer
la g´eom´etrie d’absorption de l’ac´etyl`ene sur la surface de Si(001)-(2×1), celle-ci reste incertaine. Les
r´esultats pr´esent´es dans la suite ont ´et´e obtenus en collaboration avec les ´equipes de M. N. Piancastelli
de Uppsala et W. Wurth de Hambourg et d´emontrent la puissance des techniques d’absorption des
rayons X pour ´elucider la g´eom´etrie d’adsorption de l’ac´etyl`ene.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-50-320.jpg)
![5.1. AC´ETYL`ENE 31
5.1.2 G´eom´etrie d’adsorption `a saturation
Fig. 5.2 — Spectres NEXAFS de l’ac´etyl`ene pour E normal `a la surface.
Afin d’obtenir des surfaces monodomaines, nous avons utilis´e des ´echantillons vicinaux d´esorient´es
de 5◦ ce qui permet d’obtenir des terrasses monodomaines d’environ 7 nm. Deux ´echantillons tourn´es
de 90◦ ont ´et´e utilis´es de mani`ere `a pouvoir aligner le champ ´electrique des rayons X parall`ele ou
perpendiculaires aux rang´ees de dim`eres. D’autre part, la rotation de l’´echantillon autour de l’axe
du faisceau permet d’orienter le champ ´electrique des rayons X parall`element ou perpendiculairement
`a la surface. Ainsi, toutes les orientations du champ peuvent ˆetre choisies et permettre une analyse
angulaire des spectres NEXAFS. Les d´etails exp´erimentaux peuvent ˆetre obtenus dans l’article [4] de
l’annexe.
La figure 5.2 pr´esente les spectres au seuil K du carbone de l’ac´etyl`ene lorsque le champ ´electrique
est perpendiculaire `a la surface. On note que pour les deux ´echantillons, les spectres NEXAFS sont
tr`es similaires et on distingue clairement deux structures tr`es fines `a 283.8 eV et 286.7 eV ainsi
qu’une structure plus large `a 288.4 eV. L’origine des diff´erentes structures visibles sur ces spectres
sera discut´ee par la suite. Lorsqu’on aligne le champ ´electrique avec la surface, on obtient les spectres
pr´esent´es sur la figure 5.3. Lorsque le champ est parall`ele au dim`ere (Fig.5.3(a)), on reconnaˆıt les 3
structures principales `a 283.8 eV, 286.7 eV et 288.4 eV d´ej`a visibles sur les spectres pr´ec´edents ainsi
qu’une structure beaucoup plus large vers 299 eV. Le spectre obtenu lorsque le champ ´electrique est
perpendiculaire au dim`ere (Fig.5.3(b)) pr´esente les mˆemes pics mais avec une intensit´e plus importante
pour le premier pic par rapport au fond continu `a 350 eV. La d´ependance en intensit´e des pics
avec la polarisation du champ ´electrique E, permet de comprendre l’origine des diff´erentes structures
observ´ees. Ainsi, la structure dominante dans les spectres de la figure 5.3 lorsque le champ E est
parall`ele `a la surface, provient de l’orbitale mol´eculaire de type πC−C perpendiculaire `a l’axe de
la mol´ecule et repr´esente la partie anti-liante de la triple liaison de la mol´ecule d’ac´etyl`ene. Cette
structure est peu intense lorsque le champ E est perpendiculaire `a la surface, en revanche son intensit´e
est tr`es importante lorsque le champ est parall`ele `a la surface. Cela nous indique que la mol´ecule est
adsorb´ee `a plat sur la surface, ainsi cette structure peut correspondre `a une orbitale de type py d´eriv´ee
de l’orbitale anti-liante πC−C. Le structure observ´ee `a 286.7 eV, qui est elle beaucoup plus intense
lorsque le champ est perpendiculaire `a la surface est suppos´ee provenir de l’orbitale de type pz. Cet
´etat semble donc impliqu´e dans la liaison entre la mol´ecule et le substrat de silicium, c’est pour cela
qu’elle est d´enot´ee C − Si sur les spectres NEXAFS. Alors que la mol´ecule d’ac´etyl`ene pr´esente une
hybridation de type sp, celle-ci se transforme en se liant au silicium par une re-hybridation sp2 voir
sp3. Lors d’une re-hybridation de type sp3, la mol´ecule d’ac´etyl`ene peut se lier `a quatre atomes de
silicium, en revanche dans une re-hybridation de type sp2, la mol´ecule ne se lie qu’`a deux atomes de](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-51-320.jpg)
![32 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
Fig. 5.3 — Spectres NEXAFS de l’ac´etyl`ene pour E parall`ele `a la surface.
silicium. Comme la structure associ´ee `a un ´etat π−π `a 283.8 eV est pr´esent dans toutes les g´eom´etries
mesur´ees, on consid`ere que la mol´ecule d’ac´etyl`ene poss`ede une hybridation de type sp2. Dans ce cas
les configurations d’adsorption o`u la mol´ecule est li´ee `a deux dim`eres de silicium (figure 5.1 (d) et (e))
peuvent ˆetre ´elimin´ees car elles ne pr´esentent pas de re-hybridation de type sp2. Les configurations
restantes compatibles avec les mesures NEXAFS sont les suivantes : dimerized, broken − dimer et
end − bridge. La configuration broken − dimer est ´egalement peu probable car en cassant le dim`ere,
la liaison Si-Si devient plus grande et par cons´equent, la liaison C-Si est orient´ee `a 45◦ de la surface.
Or, on observe que la structure associ´ee `a la r´esonance C-Si est beaucoup plus intense lorsque le
champ E est perpendiculaire `a la surface, indiquant que cette liaison est bien normale `a la surface
et non orient´ee `a 45◦ comme dans la configuration broken − dimer. Ainsi, seules les configurations
dimerized et end−bridge restent compatibles avec les observations faites par la technique du NEXAFS.
Avant d’essayer de discriminer les diff´erentes configurations restantes, on peut s’int´eresser aux autres
structures des spectres NEXAFS. Ainsi, la structure `a 288.4 eV qui est visible `a la fois sur la figure 5.2
et la figure 5.3 est plus marqu´ee lorsque le champ ´electrique est normal `a la surface. Cette structure
est associ´ee `a une ´etat du type σC−H, en effet, lors de l’adsorption de la mol´ecule, l’orbitale C-C anti-
liante de type π se s´epare pour former la liaison entre le silicium et le carbone mais aussi la liaison C-H
qui pointe alors vers le vide. La notation de cette structure a ´et´e reprise de donn´ees de Matsui et al.
[54] chez qui elle avait d´ej`a ´et´e mise en ´evidence. A plus haute ´energie, on peut observer une derni`ere
structure tr`es large `a 299 eV qui est visible lorsque le champ E est parall`ele ou perpendiculaire `a la
surface. La position de cette structure co¨ıncide avec l’´etat d´enot´e σC−C dans l’article de Matsui et
al.. Cette structure qui d´erive de l’orbitale anti-liante de type σ est orient´ee parall`element `a l’axe de
la mol´ecule. En effet, on peut voir sur la figure 5.2 o`u le champ est normal `a la surface, que cette
structure est peu marqu´ee alors qu’elle est plus intense sur la figure 5.3 lorsque le champ est parall`ele
`a la surface. Cela confirme que la mol´ecule d’ac´etyl`ene s’adsorbe `a plat sur la surface. D’autre part la
position en ´energie de cette structure permet d’obtenir des informations sur le type et la longueur de
la liaison C-C. En effet, la r´esonance de type σ se d´ecale vers les basses ´energies lorsque l’on passe
d’une triple liaison C-C, comme dans le cas de l’ac´etyl`ene, `a une simple liaison C-C, comme dans
le cas de l’´ethane [7]. Dans le spectre NEXAFS de l’ac´etyl`ene gazeux, la r´esonance σ se situe vers
310 eV et la distance C-C vaut 1.3 ˚A environ, en revanche, cette r´esonance se d´ecale vers 300 eV
pour l’´ethyl`ene gazeux qui poss`ede une double liaison et o`u les atomes de carbone sont distants de
1.52 ˚A et on la trouve vers 290 eV dans le cas de l’´ethane. Lorsque l’ac´etyl`ene s’adsorbe sur le silicium
en adoptant une re-hybridation de type sp2, la r´esonance σ est attendue aux environs de 300 eV alors](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-52-320.jpg)
![5.2. ETHYL`ENE 33
qu’elle devrait plutˆot se trouver vers 291 eV comme dans le cas d’une re-hybridation de type sp3. La
pr´esence de la r´esonance σ `a 299 eV confirme que la mol´ecule d’ac´etyl`ene forme une liaison de type
sp2 avec le substrat. De nouveau, les configurations p − bridge et r − bridge ne sont pas compatibles
avec cette observation. En revanche, la configuration broken − dimer, o`u la distance entre carbone
a ´et´e calcul´ee comme ´etant sensiblement la mˆeme que dans le mod`ele dimerized (respectivement
1.37 ˚A et 1.36 ˚A [55]), est compatible avec l’observation de cette r´esonance `a 299 eV. Compte tenu
de toutes les observations faites, deux configurations d’adsorption peuvent toujours ˆetre envisag´ees :
dimerized et end − bridge. La re-hybridation des mol´ecules sur le silicium ´etant tr`es similaires, il est
difficile de les distinguer. Ainsi, comme il l’a ´et´e observ´e dans d’autres ´etudes les formes dimerized
et end − bridge coexistent sur la surface de Si(001)-(2×1) [47, 48, 56, 50, 51] toutefois en analysant
l’intensit´e de la r´esonance πC−C (figure 5.3), il est possible de quantifier l’abondance de chacune des
esp`eces pr´esentes. Ainsi, la r´esonance πC−C, qui est observ´ee lorsque le champ est perpendiculaire `a
l’axe de la mol´ecule est plus intense d’environ 20% lorsque le champ est ´egalement perpendiculaire
aux dim`eres. Cela indique qu’il existe une population plus importante de mol´ecules adsorb´ees dans la
configuration dimerized avec un rapport d’occupation de 5 : 4 entre les configurations dimerized et
end − bridge.
Ainsi, la g´eom´etrie d’adsorption de l’ac´etyl`ene a pu ˆetre d´etermin´ee par NEXAFS, faisant apparaˆıtre
la co-existence de deux sites d’adsorption sur la surface. Ces r´esultats sont d´etaill´es dans l’article [4]
de l’annexe. Malheureusement, pour des raisons techniques, il n’a pas ´et´e possible d’´etudier ce syst`eme
par les techniques optiques de surface dont nous disposons `a l’INSP.
5.2 Ethyl`ene
5.2.1 Le syst`eme C2H4/Si(001)-(2×1)
Fig. 5.4 — Repr´esentation sch´ematique des configurations d’adsorption de l’´ethyl`ene.
L’´etude d’hydrocarbures sur silicium se poursuit logiquement par l’´etude de l’adsorption d’´ethyl`ene
sur Si(001) qui poss`ede une double liaison C=C. L’´etude de ce syst`eme a d´ebut´e il y a une dizaine
d’ann´ee [57], d´emontrant que la mol´ecule s’adsorbe en formant une liaison de type di-σ sur les liaisons
pendantes du silicium comme l’illustre la figure 5.4 (a). Dans cette g´eom´etrie, l’´ethyl`ene perd sa double](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-53-320.jpg)
![34 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
0.5 MC
1 MC
dimerized bridge
Fig. 5.5 — Calcul des g´eom´etries dans les configurations bridge et dimerized pour des taux de cou-
verture de 0.5 MC et 1 MC.
liaison et les atomes de carbone se re-hybrident dans un ´etat du type sp3. Sous l’effet de l’adsorption,
toutes les liaisons pendantes du silicium sont satur´ees et l’oscillation du dim`ere est supprim´ee [20].
Des ´etudes exp´erimentales ont permis de confirmer cette configuration toutefois une controverse s’est
d´evelopp´ee sur la base d’autres ´etudes qui mettent en ´evidence une configuration o`u le dim`ere de
silicium est cliv´e lors de l’adsorption menant `a la g´eom´etrie pr´esent´ee sur la figure 5.4 (c) [58, 59].
Deux ´etudes conjointes [60, 61] men´ees par photo´emission sur la bande de valence (UPS) et par calculs
utilisant la fonctionnelle de la densit´e (DFT) ont permis de proposer une configuration d’adsorption de
l’´ethyl`ene plus d´etaill´ee. Grˆace `a la photo´emission, les auteurs ont pu mettre en ´evidence la pr´esence
de deux ´etats de la bande de valence ayant un caract`ere d´elocalis´e le long de la rang´ee de dim`ere qui a
´et´e confirm´ee par des calculs DFT. Pourtant, le recouvrement d’orbitales provenant des liaisons C-H
provoque des r´epulsions de Pauli ce qui entraˆıne la rotation de l’axe C-C autour de la normale `a la
surface et la mol´ecule adopte alors la g´eom´etrie sch´ematis´ee sur la figure 5.4 (b). Ainsi, la rotation de
la mol´ecule d’un angle de 11.4◦ r´eduit la r´epulsion de Pauli et permet la formation d’´etats d´elocalis´es.
Ces nouveaux r´esultats incitent `a vouloir mieux comprendre la distribution des ´etats de valence vides
et occup´es de l’´ethyl`ene adsorb´e sur Si(001)-(2×1).
La situation concernant les calculs th´eoriques n’est ´egalement pas compl`etement clarifi´ee `a l’heure
actuelle ; en effet, une ´etude th´eorique r´ecente bas´ee sur des calculs de structure a montr´e, que la config-
uration dimerized est la plus stable pour un taux de couverture de 0.5 MC alors qu’`a saturation, c’est
la configuration bridge (voir Fig. 5.5) qui apparaˆıt ˆetre la plus stable [62, 51]. Cette diff´erence entre la
g´eom´etrie la plus stable qui est calcul´ee et celle qui est effectivement observ´ee exp´erimentalement, peut
provenir d’effet de cin´etique d’adsorption. En effet, le taux de couverture atteint durant le processus
d’adsorption et `a saturation est un param`etre important pour d´eterminer la configuration d’adsorption
des mol´ecules d’´ethyl`ene. Des images obtenues par STM montrent que les mol´ecules ont tendance `a
ne pas occuper les dim`eres directement dans le voisinage d’une mol´ecule adsorb´ee ce qui indique que
l’interaction entre mol´ecules n’est pas n´egligeable [63]. Dans ces conditions, le taux de couverture `a
saturation devrait ˆetre de 0.5 MC. Dans une autre ´etude par photo´emission sur les niveaux de cœur,
un taux de couverture de 0.87 MC est estim´e `a saturation [64]. Il apparaˆıt ´egalement selon une autre](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-54-320.jpg)
![5.2. ETHYL`ENE 35
´etude exp´erimentale par photo´emission, que le coefficient de collage chute de mani`ere dramatique pour
des taux de couverture sup´erieurs `a 0.5 MC rendant difficile la saturation de la surface [65].
Ici, la comparaison entre les spectres optiques calcul´es par des m´ethodes ab initio et les spectres
optiques mesur´es sur une surface vicinale de Si(001)-2×1 peuvent nous informer sur la g´eom´etrie d’ad-
sorption des mol´ecules d’´ethyl`ene, allant des faibles taux de couverture `a la saturation. De mani`ere
compl´ementaire, des mesures de NEXAFS sur la surface de silicium satur´ee en ´ethyl`ene, permettent de
mettre en ´evidence la distribution des orbitales mol´eculaires vides de l’´ethyl`ene adsorb´e et confirment
les r´esultats obtenus en photo´emission par Widdra et al [60] concernant la g´eom´etrie d’adsorption `a
saturation.
5.2.2 G´eom´etrie d’adsorption pour 0.5 MC et 1 MC
Cette ´etude a ´et´e r´ealis´ee en collaboration avec l’´equipe de th´eoriciens du Pr. Del Sole `a Rome,
notamment M. Marsili et O. Pulci. Les d´etails des proc´edures utilis´ees pour calculer la configuration
de l’´ethyl`ene la plus stable ainsi que l’obtention des spectres de r´eflectivit´e anisotrope sont expos´es
dans l’article [66]. Les calculs r´ev`elent que la configuration d’adsorption la plus stable pour un taux
de couverture de 0.5 MC est la configuration dimerized alors que pour un taux de recouvrement de
1 MC, c’est la configuration bridge qui du point de vue ´energ´etique est la plus stable. Ces r´esultats
sont en accord avec les r´esultats obtenus dans d’autres ´etudes pr´ec´edentes [62, 51]. Les calculs de
structure dimerized et bridge pour des taux de couverture de 0.5 MC et 1 MC sont pr´esent´es sur la
figure 5.5.
Les spectres de r´eflectivit´e anisotrope calcul´es pour 0.5 MC et 1 MC sont repr´esent´es sur les figures 5.6
(a) et (c) respectivement en consid´erant les deux g´eom´etries bridge et dimerized, alors que les spectres
exp´erimentaux pour 0.5 MC et 1 MC sont pr´esent´es sur les figures 5.6 (b) et (d). Afin de valider la
qualit´e des calculs th´eoriques, le spectre de la surface propre du silicium a ´et´e ´egalement calcul´e et est
pr´esent´e, en pointill´e, sur les deux figures th´eoriques, il peut ˆetre compar´e au spectre mesur´e sur une
surface de silicium nominale monodomaine, obtenue par ´electromigration. On remarque que l’accord
entre spectre calcul´e et spectre exp´erimental est tout `a fait satisfaisant , autant pour les grandes
´energies de photon proches du point critique E2, que pour les petites ´energies de photon comme pour
l’´etat de surface du silicium qui apparaˆıt vers 1.6 eV. Afin de pouvoir comparer spectres th´eoriques
et spectres exp´erimentaux, la contribution due aux marches a ´et´e soustraite du spectre exp´erimental
mesur´e sur 0.5 MC adsorb´e sur une surface vicinale en employant la m´ethode d´ecrite par Jaloviar et al
[13]. Le spectre r´esultant est celui pr´esent´e sur la figure 5.6 (b). Si l’on compare `a pr´esent les spectres
calcul´es pour 0.5 MC pour les configurations bridge et dimerized avec le spectre exp´erimental ne
comportant pas de contribution due aux marches, on s’aper¸coit que pour les hautes ´energies les spectres
calcul´es sont tr`es similaires et correspondent correctement `a ce qui est obtenu exp´erimentalement.
Dans ces conditions, les calculs ne permettent pas de diff´erencier les deux configurations. En revanche
pour les faibles ´energies, les deux spectres calcul´es diff`erent consid´erablement ; alors que le spectre
correspondant `a la configuration bridge pr´esente une structure n´egative tr`es marqu´ee `a 1.6 eV, le
spectre calcul´e pour la configuration dimerized est faiblement structur´e. Toutefois la comparaison
avec les spectres exp´erimentaux dans cette gamme d’´energie s’av`ere difficile, les mesures ayant ´et´e
effectu´ees sur des ´echantillons vicinaux o`u l’´etat de surface est fortement att´enu´e. Ainsi, la comparaison
entre spectres calcul´es et spectres exp´erimentaux ne permet pas de d´ecider entre l’une ou l’autre des
configurations. Dans le cas de 1 MC d’´ethyl`ene, les spectres calcul´es pour les deux configurations sont
tr`es diff´erents, on note la pr´esence principale d’une structure n´egative `a 3.8 eV pour le spectre de la
configuration bridge, alors que le spectre de la configuration dimerized pr´esente plusieurs structures,
notamment les structures A et B que l’on retrouve dans le spectre exp´erimental. On voit ici que
c’est le spectre calcul´e pour la configuration dimerized qui reproduit le plus fid`element les donn´ees](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-55-320.jpg)
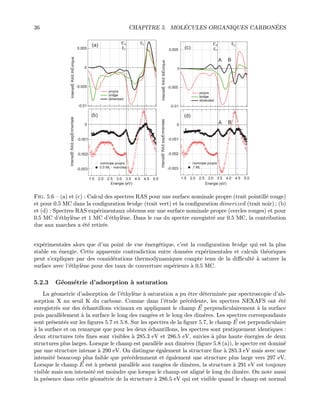
![5.2. ETHYL`ENE 37
Fig. 5.7 — Spectres NEXAFS de l’´ethyl`ene pour E normal `a la surface.
Fig. 5.8 — Spectres NEXAFS de l’´ethyl`ene pour E parall`ele `a la surface.
`a la surface ; `a plus haute ´energie, on retrouve la structure plus large vers 297 eV.
Lorsque le champ E est parall`ele aux dim`eres, le spectre de la figure 5.8 (a) est domin´e par une struc-
ture `a 291 eV. Cette structure est ´egalement visible, avec une intensit´e plus faible, lorsque le champ
est perpendiculaire aux dim`eres ; en revanche on la distingue `a peine quand le champ est normal `a la
surface. Cette structure peut donc ˆetre associ´ee `a une orbitale mol´eculaire dans le plan de la surface
et parall`ele `a l’axe de la mol´ecule, d´erivant d’une r´esonance de type σ . Alors que la structure de
type σ correspondant `a la double liaison C=C se trouve `a 301 eV dans l’´ethyl`ene gazeux [67], cette
r´esonance se d´ecale vers 291 eV lorsque la mol´ecule est adsorb´ee comme dans le cas de l’´ethane gazeux
pr´esentant une hybridation de type sp3 [67]. Le d´eplacement de cette structure vers les basses ´energies
d´emontre que la mol´ecule perd sa double liaison et se rehybride en formant une liaison de type sp3 avec
le substrat. En revanche, cette structure ´etant toujours visible quand le champ est perpendiculaire
aux dim`eres, le spectre NEXAFS montre de mani`ere univoque que la mol´ecule est l´eg`erement vrill´ee
autour de l’axe normal `a la surface. Lorsque le champ E est `a pr´esent perpendiculaire `a la surface,
deux structures fines `a 285.3 eV et 286.5 eV dominent les spectres NEXAFS des figures 5.7 (a) et (b).
Des mesures r´ealis´ees sur l’´ethyl`ene gazeux [67] ou dans des syst`emes peu li´es [68] montrent une unique
structure intense correspondant `a un ´etat de type π quand le champ est perpendiculaire `a l’axe de la
mol´ecule. En revanche ici, deux structures tr`es fines apparaissent mais avec des intensit´es plus faibles](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-57-320.jpg)
![38 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
que la r´esonance associ´ee `a la r´esonance σ de la figure 5.8 (a). Ainsi, on peut mettre en ´evidence ici
que la mol´ecule perd sa liaison de type π et se re-hybride lorsqu’elle est adsorb´ee sur le silicium. Dans
les spectres d’adsorption, nous avons acc`es aux parties anti-liantes des orbitales de type sp3, `a savoir,
la liaison C-Si dirig´ee vers le substrat, la liaison de type σ C-C et deux liaisons C-H ´equivalentes qui
pointent vers l’ext´erieur. Lorsque le champ est perpendiculaire `a la surface, seuls les ´etats sortant
de la surface pourront ˆetre sond´es, c’est `a dire ici la liaison C-Si et les liaisons C-H. Ainsi, les deux
structures fines observ´ees sur la figure 5.7 sont attribu´ees `a ces deux types de liaison mais il n’est
pas possible de d´eterminer sans ambigu¨ıt´e l’origine de chacune des structures. Des mesures NEXAFS
r´ealis´ees sur une couche ´epaisse de mol´ecules condens´ees `a 60 K [54] peuvent permettre d’assigner ces
deux structures, ainsi, la premi`ere `a 285.3 eV correspondrait `a la liaison C-Si et la seconde `a 286.5 eV
correspondrait aux liaisons C-H. Nos observations corroborent la correspondance propos´ee, ainsi la
structure `a 285.3 eV est visible lorsque le champ est parall`ele aux dim`eres mais aussi apparaˆıt comme
un ´epaulement quand le champ est perpendiculaire aux dim`eres, un tel comportement est attendu si la
mol´ecule est l´eg`erement vrill´ee autour de la normale `a la surface. La seconde r´esonance `a 286.5 eV est
alors associ´ee `a un m´elange d’´etats de type C-H2. On la retrouve lorsque le champ est perpendiculaire
aux dim`eres `a une ´energie plus grande qui peut s’expliquer par la dispersion de cet ´etat comme il a ´et´e
montr´e en photo´emission [60, 61]. Cet ´etat devrait ´egalement ˆetre visible lorsque le champ est align´e
avec le dim`ere mais compte tenu de la structure tr`es intense associ´ee de type σC−C elle n’apparaˆıt pas
clairement sur la figure 5.8 (a).
L’´etude de ce syst`eme mod`ele d’hydrocarbones sur silicium d´emontre la compl´ementarit´e des tech-
niques optiques de surface et d’absorption X pour ´elucider la g´eom´etrie d’adsorption de mol´ecules `a
diff´erents taux de couverture. Alors que des taux de couverture inf´erieurs `a 0.5 MC sont difficiles `a
´etudier par absorption X, on s’aper¸coit que les technique optiques de surface ont une bien meilleure
sensibilit´e `a la surface et peuvent renseigner sur les premiers stades de l’adsorption de mol´ecules. Les
r´esultats obtenus en optiques ont ´et´e r´ecemment soumis `a la revue Phys. Rev. B pour publication
(article [5] de l’annexe), les r´esultats concernant l’absorption X ont, quant `a eux, ´et´e publi´es dans
l’article [6] de l’annexe.
5.3 Benz`ene
5.3.1 Le syst`eme C6H6/Si(001)-(2×1)
L’´etude de l’adsorption de mol´ecules organiques carbon´ees se poursuit avec le benz`ene, qui repr´esen-
te la plus simple des mol´ecules aromatiques et qui est un syst`eme mod`ele id´eal pour la compr´ehension
des m´ecanismes r´egissant l’adsorption de cette classe de mol´ecule sur silicium. L’adsorption du benz`ene
sur la surface de silicium (001) a ´et´e largement ´etudi´ee depuis de nombreuses ann´ees autant de mani`ere
exp´erimentale que th´eorique [15, 69, 17, 70, 16, 71]. La premi`ere ´etude exp´erimentale est men´ee par
Tagushi et al. [15] en utilisant diff´erentes techniques telles que la spectroscopie Auger, la perte d’´energie
des ´electrons (HREELS) ou encore la d´esorption thermique (TDS). Cette ´etude pr´ecoce montre alors
que le benz`ene se chimisorbe sur la surface de silicium (001) `a temp´erature ambiante avec un taux
de couverture d’environ 0.25 mol´ecule par atome de silicium (un dim`ere sur deux est occup´e par une
mol´ecule de benz`ene). L’observation de deux pics de d´esorption thermique indique qu’il existe plusieurs
modes d’adsorption possibles associ´es `a l’adsorption sur les terrasses et l’adsorption sur les marches.
Les mesures HREELS r´ev`elent la pr´esence d’une double liaison C=C, les auteurs proposent alors deux
mod`eles d’adsorption possibles pr´esent´es sur la figure 5.9 (a) et (b), le mod`ele 1,4-cycloh´exadi`ene ou
“butterfly” et le mod`ele 1,3-cycloh´exadi`ene ou “tilted” o`u la mol´ecule de benz`ene s’adsorbe sur le
silicium en formant une liaison de type sp3. Plusieurs ´etudes th´eoriques ont alors ´et´e men´ees pour
savoir quelle est la configuration qui est la plus stable. Ainsi pour Craig [69] c’est la configuration](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-58-320.jpg)
![5.3. BENZ`ENE 39
Fig. 5.9 — Repr´esentation sch´ematique des g´eom´etries d’adsorption du benz`ene.
“tilted” qui est la plus stable alors que pour Jeong et al. [72] c’est une autre configuration o`u le
benz`ene est `a plat sur la surface qui est pr´ef´er´e (voir figure 5.9 (c)). Dans cette nouvelle configuration
nomm´ee “pedestal”, le benz`ene se trouve li´e `a deux dim`eres adjacents et perd la double liaison C=C.
R´ecemment une ´etude men´ee par photo´emission r´ev`ele une adsorption de type “butterfly” [70], alors
que deux ´etudes en STM ind´ependantes mettent en ´evidence que l’adsorption initiale du benz`ene
dans la configuration “butterfly” est m´etastable et que les mol´ecules basculent dans une configuration
plus stable de type “tilted” [16] ou “pedestal” [17] selon l’´etude. Cette id´ee est aussi support´ee par
une ´etude men´ee en NEXAFS o`u une seconde g´eom´etrie d’adsorption de type “pedestal” est mise en
´evidence [71].
Afin de clarifier la g´eom´etrie d’adsorption du benz`ene sur Si(001)-(2×1), une ´etude NEXAFS r´esolue
angulairement a ´et´e entreprise sur des ´echantillons monodomaines et montre clairement l’adsorption
d’une seule esp`ece sur la surface. D’autre part, le changement de conformation sugg´er´e par les deux
´etudes STM pr´ec´edemment cit´ees, a pu ˆetre ´etudi´e grˆace `a la technique RAS.
5.3.2 G´eom´etrie d’adsorption
La figure 5.10 pr´esente les spectres NEXAFS enregistr´es lorsque le champ est perpendiculaire `a la
surface. Dans cette g´eom´etrie on remarque que le spectre est domin´e par une structure fine `a 284.8 eV,
on distingue ´egalement la pr´esence d’une petite structure `a 286.9 eV. Lorsque le champ est parall`ele
`a la surface, les spectres obtenus sont tr`es diff´erents (voir Fig. 5.11), les deux pr´ec´edentes structures
sont toujours visibles avec des intensit´es diff´erentes et deux structures plus larges apparaissent vers
292.5 eV et 300.7 eV. La structure intense `a 284.8 eV qui domine les spectres de la figure 5.10 poss`ede
un caract`ere π∗, cela indique que cet ´etat poss`ede une grande contribution hors du plan comme dans le
cas de l’adsorption sur les m´etaux [73]. Toutefois, si la mol´ecule s’adsorbe `a plat sur le silicium, cette
structure disparaˆıt des spectres lorsque le champ ´electrique est parall`ele `a la surface. Or, il est clair sur
la figure 5.11(b) que la structure correspondant aux ´etats de type π∗ est toujours pr´esente et donc que
la mol´ecule n’est pas compl`etement `a plat sur la surface. D’autre part, cette structure est beaucoup
plus marqu´ee lorsque le champ est perpendiculaire au dim`ere. Cette observation est compatible avec](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-59-320.jpg)
![40 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
Fig. 5.10 — Spectres NEXAFS du benz`ene pour E perpendiculaire `a la surface.
les configurations d’adsorption de type “butterfly” ou “tilted”. Dans une pr´ec´edente ´etude men´ee en
NEXAFS [71], la structure associ´ee aux ´etats de type π∗ est asym´etrique en raison de la pr´esence d’une
deuxi`eme structure d’environ 1 eV plus ´elev´ee, les auteurs attribuent cette asym´etrie `a la pr´esence
d’une deuxi`eme configuration d’adsorption. Compte tenu de la r´esolution utilis´ee ici de 50 meV, une
telle structure aurait ´et´e clairement r´esolue, ce qui n’est pas le cas. Ainsi, dans notre syst`eme il n’y
a pas d’´evidence d’adsorption d’une deuxi`eme esp`ece sur la surface. La seconde structure `a 286.9 eV
a ´et´e attribu´ee dans une pr´ec´edente ´etude aux ´etats du type C-H∗. Ainsi, dans les trois polarisations
propos´ees, cette structure poss`ede une intensit´e similaire ce qui indique que les ´etats C-H∗ ne sont
ni parall`eles, ni perpendiculaires `a la surface mais pointent vers l’ext´erieur comme il est propos´e
dans les trois mod`eles “butterfly”, “tilted” et “pedestal”. Dans les spectres NEXAFS pr´esent´es, on
ne retrouve pas de structure vers 289 eV qui correspondrait `a la conjugaison des ´etats de type π∗
comme on peut le voir sur les spectres obtenus en phase gazeuse ou sur des multicouches [73, 74].
Cela indique que le syst`eme aromatique π a ´et´e d´etruit lors de l’adsorption du benz`ene sur le silicium.
Si on regarde maintenant `a plus haute ´energie lorsque la polarisation est parall`ele aux dim`eres, deux
structures plutˆot larges sont visibles `a 292.5 eV et 300.7 eV. La structure de plus basse ´energie peut
ˆetre associ´ee `a une r´esonance de type σ∗
C−C entre les atomes de carbone du benz`ene avec une ´energie
Fig. 5.11 — Spectres NEXAFS du benz`ene pour E parall`ele `a la surface.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-60-320.jpg)
![5.3. BENZ`ENE 41
plus faible que dans le cas du benz`ene en phase gazeuse en raison de l’´elongation des liaisons simples
C-C lors de l’adsorption [61]. La pr´esence de cette structure lorsque le champ est perpendiculaire aux
dim`eres indique que les liaisons simples C-C sont plutˆot sym´etriques par rapport `a l’axe du dim`ere
comme dans le mod`ele “butterfly”. Dans le mod`ele “tilted” les liaisons simples C-C sont parall`eles
aux dim`eres, on s’attend alors `a observer dans ce cas une structure associ´ee aux r´esonances σ∗
C−C plus
marqu´ee lorsque le champ est parall`ele aux dim`eres. Cette observation favorise la g´eom´etrie de type
“butterfly”. La seconde structure `a 300.7 eV sur la figure 5.11(a) peut ˆetre associ´ee aux r´esonances
de type σ∗
C=C de la double liaison C=C, avec une longueur plus petite que dans le cas du benz`ene
gazeux [61]. La pr´esence d’un double liaison C=C a ´et´e mise en ´evidence par des mesures HREELS
[75], l’observation d’une structure associ´ee `a une double liaison invalide le mod`ele “pedestal” qui ne
poss`ede que de simples liaisons C-C, il reste alors deux mod`eles compatibles : “butterfly” et “tilted”.
Dans le spectre de la figure 5.11(b), une structure `a 297 eV est visible alors qu’`a 300.7 eV un vague
´epaulement subsiste. Si on associe cette structure `a une r´esonance de type σ∗
C=C, cela voudrait dire
que cette double liaison qui est visible pour deux directions de polarisation n’est align´ee ni selon le
dim`ere, ni selon la rang´ee de dim`ere ce qui favorise le mod`ele “tilted”. Toutefois, si cette structure
de la figure 5.11(b) `a 297 eV correspond `a une double liaison, la longueur de cette double liaison
est encore plus courte que dans le cas pr´ec´edent ce qui n’est pas compatible avec le mod`ele “tilted”.
Ainsi, il est difficile de donner une origine `a la pr´esence de cette structure `a 297 eV qui est visible
lorsque le champ est perpendiculaire aux dim`eres. Ainsi, l’assignement des structures NEXAFS n’est
pas toujours direct et la mise en jeux d’hybridations d’orbitales peut compliquer le spectre observ´e.
Toutefois, l’ensemble des observations d´eduites des spectres NEXAFS enregistr´es sur les surfaces de
silicium (001) satur´ees en benz`ene conduisent `a pr´ef´erer le mod`ele “butterfly” comme configuration
d’adsorption `a temp´erature ambiante. Ces r´esultats sont d´etaill´es dans l’article [7] de l’annexe.
5.3.3 Changement de conformation
Malgr´e les nombreuses ´etudes men´ees par le pass´e sur l’adsorption du benz`ene sur Si(100), des
interrogations subsistent en ce qui concerne le possible changement de conformation de la mol´ecule
avec le temps. En effet, deux ´etudes ind´ependantes en microscopie `a effet tunnel ont montr´e que
Fig. 5.12 — Repr´esentation de l’´evolution
des spectres de RA sur une surface de
Si(001) satur´ee en benz`ene en fonction
du temps. Les variations du signal sont
rep´esent´ees par les niveaux de couleurs.
(a)
(b)
-0.0010
-0.0005
0.0000
0.0005
Re(∆r/r)
5.55.04.54.03.53.02.5
Energie (eV)
14h après saturation
silicium propre après 1 nuit
-0.0010
-0.0005
0.0000
0.0005
Re(∆r/r)
6 h après l'adsorption
benzène contaminé
Fig. 5.13 — Comparaison des spectres de RA de
C6H6/Si(001) obtenus apr`es l’adsorption et des spectres
de RA sur des surfaces contamin´ees.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-61-320.jpg)
![42 CHAPITRE 5. MOL´ECULES ORGANIQUES CARBON´EES
Re(∆r/r)
5.04.54.03.53.02.52.0
Energie (eV)
0.5x10
-3
(a)
(b)
(c)
(d)
Si(001) propre
C6H6/Si(001)
C6H6/Si(001) après 5 min @ 120∞C
C6H6/Si(001) après 5 min @ 200∞C
Fig. 5.14 — Spectres de RA : (a) : surface satur´ee en benz`ene, (b) : surface satur´ee en benz`ene apr`es
recuit 5min `a 120◦C, (c) : surface satur´ee en benz`ene apr`es recuit 5 min `a 200◦C, (d) : surface Si(001)
propre.
la mol´ecule s’adsorbe initialement dans une conformation m´etastable de type “butterfly” et bascule
dans une seconde g´eom´etrie plus stable de type “pedestal” [16] ou “tilted” [17]. Des mesures par
spectroscopie optique anisotrope ont ´et´e men´ees afin de savoir si il ´etait possible ou non de mettre en
´evidence un tel changement de conformation. Une ´etude pr´eliminaire d´ecrite dans la section 3.3.3 a
montr´e que les techniques RAS et SRDS peuvent donner des informations quantitatives sur le nombre
de dim`eres qui a r´eagi lors de l’adsorption. Cette propri´et´e a ´et´e exploit´ee pour essayer d’´elucider le
changement possible de conformation des mol´ecules de benz`ene. Ainsi, nous avons pu montrer par
les deux techniques optiques qu’un peu moins d’un dim`ere sur deux est occup´e par une mol´ecule de
benz`ene, ce qui est compatible avec les mod`eles “butterfly” et “tilted”. Pour mettre en ´evidence un
possible changement de conformation, une surface vicinale de Si(100) a ´et´e satur´ee en benz`ene et
laiss´ee toute la nuit dans l’enceinte ultra vide. Toutes les 15 minutes environ, un spectre de r´eflectivit´e
anisotrope s’enregistrait automatiquement. La figure 5.12 rassemble l’ensemble des spectres de RA en
fonction de l’´energie et du temps, le signal Re(∆r
r ) est traduit en niveau de couleur. On s’aper¸coit sur
cette figure que le spectre de RA se modifie tr`es rapidement avec le temps, et au bout de 6 heures
environ, les spectres n’´evoluent plus. On pourrait alors croire que l’on a ainsi mis en ´evidence une
modification de la g´eom´etrie d’adsorption du benz`ene avec le temps. On va voir qu’il n’en est rien. En
effet, d’apr`es les travaux effectu´e dans l’´equipe de Lopinski et al. [16], 85% des mol´ecules de benz`ene
se sont converties au bout de 150 minutes. On remarque dans notre cas, une variation beaucoup plus
lente ce qui traduirait une vitesse plus lente de conversion des mol´ecules de benz`ene. Si on regarde
maintenant plus en d´etail la forme des spectres de RA pr´esent´es sur la figure 5.13, on voit que le
spectre obtenu 6 heures apr`es l’adsorption est tr`es similaire au spectre obtenu lorsque le benz`ene est
contamin´e par de l’eau. De plus, le spectre obtenu 14 heures apr`es l’adsorption est ´egalement tr`es
similaire `a celui obtenu sur une surface propre de silicium laiss´ee une nuit dans l’enceinte ultravide.
On peut alors ´emettre des doutes quant `a l’origine de l’´evolution du spectre de r´eflectivit´e anisotrope](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-62-320.jpg)
![5.3. BENZ`ENE 43
de la surface satur´ee par du benz`ene avec le temps. Pour essayer d’activer thermiquement la conversion
des mol´ecules de benz`ene, une surface de benz`ene a ´et´e pr´epar´ee et chauff´ee `a environ 120◦C pendant
5 minutes. Le spectre obtenu apr`es le chauffage (figure 5.14(b)) est n’a pas ´evolu´e, nous avons alors
chauff´e le mˆeme ´echantillon `a 200◦C pendant 5 minutes. Le spectre apr`es ce deuxi`eme chauffage (figure
5.14(c)), qui ressemble au spectre de silicium avant exposition au benz`ene (figure 5.14(d)), montre que
les mol´ecules de benz`ene ont ´et´e d´esorb´ees. Ainsi, aucune activation thermique n’a pu ˆetre d´emontr´ee
o`u les mol´ecules de benz`ene transiteraient d’une configuration de type “butterfly”, o`u la mol´ecule est
li´ee `a un seul dim`ere de silicium, `a une configuration de type “pedestal”, o`u la mol´ecule de benz`ene
est adsorb´ee `a plat sur la surface en se liant `a deux dim`eres adjacents. Ainsi, la technique RAS a ´et´e
d´eterminante pour corroborer les ´etudes NEXAFS et d´emontrer une adsorption d´efinitive du benz`ene
dans une configuration de type “butterfly”. Ces r´esultats sont d´etaill´es dans l’article [8] de l’annexe.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-63-320.jpg)

![Conclusion
L’ensemble des r´esultats pr´esent´es dans ce manuscrit d´emontre les possibilit´es offertes par les
techniques de spectroscopies optiques de surface (RAS et SRDS) ainsi que la puissance des tech-
niques d’absorption X au voisinage des seuils (NEXAFS) pour caract´eriser l’adsorption d’atomes ou
de mol´ecules plus complexes sur la surface Si(001)-(2×1).
Avant de caract´eriser les adsorbats, un proc´ed´e efficace pour pr´eparer les surfaces monodomaines de
silicium, que ce soit vicinales ou nominales, a dˆu ˆetre mis en place. L’utilisation de techniques de
surface compl´ementaires telles que la diffraction d’´electrons lents ou la microscopie `a effet tunnel nous
a permis de mieux comprendre la r´eponse optique de la surface et d’utiliser la RAS pour caract´eriser
la qualit´e de la surface propre. La RAS se r´ev`ele ˆetre une technique id´eale pour quantifier la propor-
tion des deux domaines d’orientation des dim`eres qui peuvent co-exister sur les surfaces suppos´ees
monodomaines. Une fois, cette ´etape franchie, les techniques RAS et SRDS ont pu ˆetre utilis´ees pour
´etudier l’adsorption dans le cas de syst`emes mod`eles : H/Si(001)-(2×1) et O2/Si(001)-(2×1). Dans le
premier syst`eme, ´etudi´e depuis de tr`es nombreuses ann´ees, il a ´et´e possible de mettre en ´evidence le
clivage des dim`eres de silicium lors de l’adsorption de l’hydrog`ene atomique `a temp´erature ambiante
en utilisant la technique SRDS. Ainsi, la r´eponse optique caract´eristique d’une surface, o`u tous les
dim`eres ont ´et´e cass´ees, a ´et´e isol´ee et le spectre optique correspondant repr´esente une signature de
r´ef´erence. Cela constitue un r´esultat important tant il est ais´e de suivre l’´evolution du signal SRDS au
cours de l’adsorption de mol´ecules et de savoir si les dim`eres ont subi un clivage ou au contraire sont
rest´es intacts. Ce r´esultat a ´et´e utilis´e avec succ`es pour ´etudier l’adsorption de ph´enylac´etyl`ene sur
Si(001)-(2×1) et a permis de d´emontrer, avec l’aide de mesures par STM, que plusieurs sites d’adsorp-
tion doivent ˆetre envisag´es mais que n´eanmoins les dim`eres restent intacts au cours de l’adsorption
(article [1] de la liste de publications). Le second syst`eme mod`ele O2/Si(001)-(2×1), qui revˆet une
importance primordiale dans la fabrication de syst`emes micro´electroniques, nous a permis de montrer
que lorsque des calculs ab initio sont utilis´es pour calculer la r´eponse optique de la surface avec ad-
sorbat, il est possible d’extraire des informations sur le lieu d’incorporation des atomes d’oxyg`ene et
les m´ecanismes r´eactionnels mis en jeu lors de l’exposition.
Des syst`emes plus complexes faisant intervenir plusieurs types d’atomes (carbone et hydrog`ene) ont
ensuite ´et´e ´etudi´es, ainsi les modes d’adsorption de l’ac´etyl`ene, de l’´ethyl`ene ou encore du benz`ene ont
pu ˆetre d´etermin´es avec pr´ecision grˆace `a la technique du NEXAFS. On a pu montrer que l’ac´etyl`ene
s’adsorbe selon deux g´eom´etries parall`eles ou perpendiculaires aux dim`eres de silicium, que l’´ethyl`ene
`a saturation s’adsorbe sur les dim`eres de silicium et que le benz`ene s’adsorbe un dim`ere sur deux dans
une configuration dite “butterfly” sur les dim`eres de silicium. Cette technique apparaˆıt incontournable
pour comprendre comment les mol´ecules se rehybrident en arrivant sur la surface de Si(001)-(2×1).
Elle a en outre ´et´e utilis´ee r´ecemment pour ´etudier l’adsorption de la pyridine, mol´ecule cyclique
poss´edant un atome d’azote qui en s’adsorbant sur les dim`eres de silicium peut cr´eer une liaison da-
tive Si :N. Avec ces syst`emes plus complexes, on a pu ´egalement d´emontrer que les techniques optiques
seules ou compar´ees `a des calculs ab initio permettent ´egalement d’obtenir des informations sur la
g´eom´etrie d’adsorption des mol´ecules. Ainsi, les tr`es bons accords entre spectres RAS mesur´es pour
45](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-65-320.jpg)









![Bibliographie
[1] D. E. Aspnes, J. P. Harbison, A. A. Studna, and L. T. Florez. J. Vac. Sci. technol. A, 6, 1327,
1988.
[2] F. Manghi, R. Del Sole, A. Selloni, and E. Molinari. Phys. Rev. B, 41, 9935, 1990.
[3] R. Denecke, P. V¨aterlein, M. B¨assler, N. Wassdahl, A. Nilsson, J. E. Rubensson, J. Nordgren,
N. M˚artensson, and R. Nyholm. J. El. Spec. Rel. Phen., 101-103, 971, 1999.
[4] L. S. Johansson, R. I. G. Uhrberg, P. Martesson, and G. W. Hansson. Phys. Rev. B, 42, 1305,
1990.
[5] A. Incze, R. Del Sole, and G. Onida. Phys. Rev. B, 71, 035350, 2005.
[6] R. Shioda and J. van der Weide. Phys. Rev. B, 57, R6823, 1998.
[7] S. Stoyanov. Appl. Surf. Sci., 60-61, 55, 1992.
[8] K. Hata, T. Kimura, S. Ozawa, and H. Shigekawa. J. Vac. Sci. Technol. A, 18, 1933, 2000.
[9] M. Palummo, G. Onida, R. Del Sole, and B. Mendoza. Phys. Rev. B, 60, 2522, 1999.
[10] W. G. Schmidt, F. Bechstedt, and J. Bernholc. Phys. Rev. B, 63, 045322, 2001.
[11] K. Hingerl, R. E. Balderas-Navarro, A. Bonanni, P. Tichopadek, and W. G. Schmidt. App. Surf.
Sci., 175-176, 769, 2001.
[12] L. Mantese, U. Rossow, and D. E. Aspnes. Appl. Surf. Sci., 107, 35, 1996.
[13] S. G. Jaloviar, J. L. Lin, F. Liu, V. Zielasek, M. McCaughan, and M. G. Lagally. Phys. Rev.
Lett., 82, 791, 1999.
[14] B. S. Swartzentruber, N. Kitamura, M. G. Lagally, and M. B. Webb. Phys. Rev. B, 47, 13432,
1993.
[15] Y. Taguchi, M. Fujisama, T. Takaoka, T. Okada, and M. Nishijima. J. Chem. Phys., 95, 6870,
1991.
[16] G. P. Lopinski, T. M. Fortier, D. J. Moffatt, and R. A. Wolkow. J. Vac. Sci. Technol. A, 16, 1037,
1998.
[17] B. Borovsky, M. Krueger, and E. Ganz. Phys. Rev. B, 57, R4269, 1998.
[18] J.J. Boland. Adv. Phys., 42, 129, 1993.
[19] R. J. Hamers, S. K. Coulter, M. D. Ellison, J. S. Hovis, D. F. Padowitz, and M. P. Schwartz. Acc.
Chem. Res., 33, 617, 2000.
[20] P. Baumg¨artel, R. Lindsay, O. Scha, T. Giessel, R. Terborg, J. T. Hoeft, M. Polcik, A. M.
Bradshaw, M. Carbone, M. N. Piancastelli, R. Zanoni, and R. L. Toomes. New J. Phys., 1, 20,
1999.
[21] M. Nagao, Y. Yamashita, S. Machida, K. Hamaguchi, F. Yasui, K. Mukai, and J. Yoshinobu.
Surf. Sci., 513, 413, 2002.
55](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-75-320.jpg)
![56 BIBLIOGRAPHIE
[22] W. G. Schmidt, S. Glutsch, P. H. Hahn, and F. Bechstedt. Phys. Rev. B, 67, 085307, 2003.
[23] L. C. Ciacchi and M. C. Payne. Phys. Rev. Lett., 95, 196101, 2005.
[24] H. Watanabe, K. Kato, T. Uda, K. Fujita, M. Ichikawa, T. Kawamura, , and K. Terakura. Phys.
Rev. Lett., 80, 345, 1998.
[25] K. Nakajima, Y. Okazaki, and K. Kimura. Phys. Rev. B, 63, 113314, 2001.
[26] S. Dreiner, M. Sch¨urmann, and C. Westphal. Phys. Rev. Lett., 93, 126101, 2004.
[27] T. W. Pi, J. F. Wen, C. P. Ouyang, R. T. Wu, , and G. K. Wertheim. Surf. Sci., 478, L333, 2001.
[28] A. Yoshigoe and Y. Teraoka. Surf. Sci., 32 verifier, 690, 2003.
[29] H. Ikegami, K. Ohmori, H. Ikeda, H. Iwano, S. Zaima, and Y. Yasuda. Jpn. J. Appl. Phys.,
35, 1593, 1996.
[30] H. Itoh, K. Nakamura, A. Kurokawa, and S. Ichimura. Surf. Sci., 482, 114, 2001.
[31] T. Uchiyama, T. Uda, and K. Terakura. Surf. Sci., 433, 896, 1999.
[32] K. Kato and T. Uda. Phys. Rev. B, 62, 15978, 2000.
[33] N. Richard, A. Est`eve, and M. Djafari-Rouhani. Comput. Mat. Sci., 33, 26, 2005.
[34] Y.J. Chabal, K. Raghavachari, X. Zhang, and E. Garfunkel. Phys. Rev. B, 66, 161315(R), 2002.
[35] Y. Widjaja and C. B. Musgrave. J. Chem. Phys., 116, 5774, 2002.
[36] T. Yasuda, L. Mantese, U. Rossow, and D. E. Aspnes. Phys. Rev. Lett., 74, 3431, 1995.
[37] F. Fuchs, W. G. Schmidt, and F. Bechstedt. Phys. Rev. B, 72, 075353, 2005.
[38] J. R. Power, O. Pulci, A. I. Shkrebtii, S. Galata, K. Hinrichs A. Astropekakis, N. Esser, R. Del
Sole, , and W. Richter. Phys.rev. B, 67, 115315, 2003.
[39] M. Nishijima, J. Yoshinobou, H. Tsuda, and M. Onchi. Surf. Sci., 192, 383, 1987.
[40] P. A. Taylor, R. M. Wallace, C. C. Cheng, W. H. Weinberg, M. J. Dresser, and W. J. Choyke. J.
Am. Chem. Soc., 114, 6754, 1992.
[41] C. Huang, W. Widdra, X. S. Wang, and W. H. Weinberg. J. Vac. Sci. Technol. A, 11, 2250, 1993.
[42] B. I. Craig and P. V. Smith. Surf. Sci., 276, 174, 1992.
[43] C. S. Carmer, B. Weiner, and M. Frenklach. J. Chem. Phys., 99, 1356, 1993.
[44] W. A. Hofer, A. J. Fisher, and R. A. Wolkow. Surf. Sci., 475, 83, 2001.
[45] W. Widdra, C. Huang, S. I. Yi, and W. H. Weinberg. J. Chem. Phys., 105, 5605, 1996.
[46] S. H. Xu, M. Keefe, Y. Yang, C. Chen, M. Yu, G. J. Lapeyre, E. Rotenberg, J. Denlinger, and
J. T. Yates. Phys. Rev. Lett., 84, 939, 2000.
[47] R. Terborg, M. Polcik, J. T. Hoeft, M. Kittel, D. I. Sayago, R. L. Toomes, and D. P. Woodruff.
Phys. Rev. B, 66, 085333, 2002.
[48] Y. Morikawa. Phys. Rev. B, 63, 033405, 2001.
[49] D. C. Sorescu and K. D. Jordan. J. Phys. Chem. B, 104, 8259, 2000.
[50] P. L. Silvestrelli, O. Pulci, M. Palummo, R. Del Sole, and F. Ancilotto. Phys. Rev. B, 68, 235306,
2003.
[51] J. H. Cho and L. Kleinman. Phys. Rev. B, 69, 075303, 2004.
[52] H. W. Yeom, S. Y. Baek, J. W. Kim, H. S. Lee, and H. Koh. Phys. Rev. B, 66, 115308, 2002.
[53] F. Matsui, H. W. Yeom, A. Imanishi, K. Isawa, I. Matsuda, and T. Ohta. Surf. Sci., 401, L413,
1998.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-76-320.jpg)
![BIBLIOGRAPHIE 57
[54] F. Matsui, H. W. Yeom, I. Matsuda, and T. Ohta. Phys. Rev. B, 62, 5036, 1998.
[55] P. L. Silvestrelli, F. Toigo, and F. Ancilotto. J. Chem. Phys., 114, 8539, 2001.
[56] W. Kim, H. Kim, G. Lee, Y. K. Hong, K. Lee, C. Hwang, D. H. Kim, and J. Y. Koo. Phys. Rev.
B, 64, 193313, 2001.
[57] J. Yoshinobu, H. Tsuda, M. Onchi, and M. Nishijima. J. Chem. Phys., 87, 7332, 1987.
[58] C. Huang, W. Widdra, and W. H. Weinberg. Surf. Sci., 315, L953, 1994.
[59] W. Widdra, S. I. Yi, R. Maboudian, G. A. D. Briggs, and W. H. Weinberg. Phys. Rev. Lett.,
74, 2074, 1995.
[60] W. Widdra, A. Fink, S. Gokhale, P. Trischberger, U. Gutdeutsch, U. Birkenheuer, N. R¨osch, and
D. Menzel. Phys. Rev. Lett., 80, 4269, 1998.
[61] U. Birkenheuer, U. Gutdeutsch, N. R¨osch, A. Fink, S. Gokhale, P. Trischberger, D. Menzel, and
W. Widdra. J. Chem. Phys., 108, 9868, 1998.
[62] J. H. Cho, L. Kleinman, C. T. Chan, and K. S. Kim. Phys. Rev. B, 63, 073306, 2001.
[63] A. J. Mayne, A. R. Avery, J. Knall, T. S. Jones, G. A. D. Briggs, and W. H. Weinberg. Surf.
Sci., 284, 247, 1993.
[64] A. Fink, W. Widdra, W. Wurth, C. Keller, M. Stichler, A. Achleitner, G. Comelli, S. Lizzit,
A. Baraldi, and D. Menzel. Phys. Rev. B, 64, 045308, 2001.
[65] F. Rochet, F. Jolly, F. Boumel, G. Dufour, F. Sirotti, and J. L. Cantin. Phys. Rev. B, 58, 11029,
1998.
[66] M. Marsili, N. Witkowski, O. Pulci, O. Pluchery, P. L. Silvestrelli, R. Del Sole, and Y. Borensztein.
Submitted Phys. Rev. B.
[67] R. McLaren, S. A. C. Clark, I. Ishi, and A. P. Hitchcock. Phys. Rev. A, 36, 1683, 1987.
[68] H. Rabus, D. Arvanitis, M. Dohmke, and K. Baberschke. J. Chem. Phys., 96, 1560, 1992.
[69] B. I. Craig. Surf. Sci., 280, L279, 1993.
[70] S. Gokhale, P. Trischberger, D. Menzel, W. Widdra, H. Droge, H.-P. Steinr¨uck, U. Birkenheuer,
U. Gutdeutsch, and N. R¨osch. J. Chem. Phys., 108, 5554, 1998.
[71] M. J. Kong, A. V. Teplyakov, J. G. Lyubovitsky, and S. F. Bent. Surf. Sci., 411, 286, 1998.
[72] H. D. Jeong, S. Ryu, Y. S. Lee, and S. Kim. Surf. Sci., 344, L1226, 1995.
[73] J. L. solomon, R. J. Madix, and J. St¨ohr. Surf. Sci., 255, 12, 1991.
[74] A. P. Hitchcock and I. Ishii. J. Electron Spectrosc. Relat. Phenom., 42, 11, 1987.
[75] R. J. Hamers, J. S. Hovis, S. Lee, H. Liu, and J. Shan. J. Phys. Chem. B, 101, 1490, 1997.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-77-320.jpg)



![ANNEXE1
RAS: An efficient probe to characterize Si(001)-(2 · 1) surfaces
N. Witkowski *, R. Coustel, O. Pluchery, Y. Borensztein
Insitute of Nano-Sciences of Paris, UMR CNRS 7588, Universities Paris VI and Paris VII, 140 Rue de Lourmel, F-75015 Paris, France
Received 26 April 2006; accepted for publication 31 August 2006
Available online 27 September 2006
Abstract
A complete inspection of the capabilities of reflectance anisotropy spectroscopy (RAS) in studying the adsorption of molecules or
atoms on the Si(001)-(2 · 1) surface is presented. First, a direct comparison between RA spectra recorded on the clean Si(001)-
(2 · 1) and the corresponding topography of the surface obtained using scanning tunneling microscopy (STM) allows us to quantify
the mixing of the two domains that are present on the surface. Characteristic RA spectra recorded for oxygen, hydrogen, water, ethylene,
benzene are compared to try to elucidate the origin of the optical structures. Quantitative and qualitative information can be obtained
with RAS on the kinetics of adsorption, by monitoring the RA signal at a given energy versus the dose of adsorbate; two examples are
presented: H2/Si(001) and C6H6/Si(001). Very different behaviours in the adsorption processes are observed, making of this technique a
versatile tool for further investigations of kinetics.
Ó 2006 Elsevier B.V. All rights reserved.
Keywords: Reflectance anisotropy spectroscopy; Scanning tunneling microscopy; Si(001); Adsorbate
1. Introduction
As the demand for new organic systems compatible with
the electronic devices is increasing, many research groups
study the chemisorptions of molecules on Si(001)-(2 · 1)
surfaces [1–6]. Reflectance Anisotropy Spectroscopy has
demonstrated its capabilities to investigate growth pro-
cesses in II–VI or III–V compounds [7,8]. The RAS is a
non-destructive surface sensitive probe that can be used
in a large variety of environments [9–11]. It measures the
difference of reflectance when the orientation of electric
field is parallel or perpendicular to the principal axes of
the surface. On the Si(001)-(2 · 1) surface, the anisotropy
of the surface is due to the presence of silicon dimers that
are formed to minimize the surface energy. On a nominal
Si(001) surface, 1 · 2 and 2 · 1 reconstructed domains
are present with an equal ratio; the two domains display sil-
icon dimer rows that are oriented at 90° with respect to
each other. Under these conditions, the RA spectrum from
one domain cancels the RA spectrum from the other one,
leading to a zero average anisotropy. To obtain a non-null
RA signal it is necessary to use surfaces that have an excess
of one of the two domains. This can be achieved by using
vicinal surfaces where single domain terraces are separated
by double-steps or by using single domain nominal surfaces
resulting from steps electromigration. Under these condi-
tions, the RAS technique appears to be suitable to charac-
terize the ratio of the two domains that are present on the
Si(001) surfaces. Considerable efforts have been made by
experimentalists and theoreticians in order to understand
the origin of the optical spectral features [12–16] that are
visible in the RA spectra of clean silicon, but the interpre-
tation of the optical changes during the adsorption of mol-
ecules or atoms remain unclear.
In this article, we want to demonstrate the ability of
RAS to quantify the ratio of the two domains on vicinal
surfaces and the capabilities of that technique to study
adsorbates on silicon. A set of different molecules or atoms
allows us to better understand the origin of the structures
that are visible in the optical spectra; next we show how
0039-6028/$ - see front matter Ó 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.susc.2006.08.045
*
Corresponding author. Tel.: +33 144274342; fax: +33 144273982.
E-mail address: Nadine.Witkowski@insp.jussieu.fr (N. Witkowski).
www.elsevier.com/locate/susc
Surface Science 600 (2006) 5142–5149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-81-320.jpg)
![this technique is sensitive to the progressive contamination
of the clean silicon surface. Finally, RAS can be used to
study the kinetics of adsorption of molecules or atoms onto
the surface.
2. Experimental details
The experiments were carried out in an ultra-high vac-
uum (UHV) preparation chamber with a base pressure of
5 · 10À11
Torr, equipped with a low energy electron diffrac-
tion apparatus (LEED) and connected to a chamber
equipped with a commercial scanning tunneling microscope
(STM) from Omicron. The Si(001) samples were vicinal
surfaces with either a 4°-miscut angle or a 2°-miscut angle
towards the [110] direction, or nominal surfaces with a max-
imum of 0.05° of disorientation. After heating at 650 °C
during one night by direct-current heating, the two vicinal
samples were flashed at 1050 °C for few seconds to remove
the oxide layer. Three flashes were necessary to ensure the
total removal of the oxide layer and get a maximum of
anisotropic signal. No cautions were used to cool down
the sample since the crucial point to obtain a surface free
of defects, is the pressure during the annealing [17]. The sin-
gle domain nominal surface is obtained by gradually heat-
ing the sample to 1050 °C by a direct current through the
sample along the [110] direction. The sample was kept at
1050 °C for several hours and the pressure never exceeded
5 · 10À10
Torr, following the procedure described by Shi-
oda et al. [18]. Ethylene, hydrogen and oxygen, stocked in
high pressure bottles, have been introduced into a gas inlet
and liquid benzene and water have been purified by repeat-
ing freeze-pump-thaw cycles before introduction into the
gas inlet. The molecules were introduced in the UHV cham-
ber through a leak valve and the residual contamination
was checked by using a mass-spectrometer.The monohy-
dride and dihydride surfaces were obtained after exposure
of the surfaces to atomic hydrogen at 305–315 °C and room
temperature respectively. The H atoms were generated by
dissociating H2 in the 10À6
Torr range with a hot W
filament. The RAS measurements were performed on a
home-made apparatus similar to the spectrometer devel-
oped by Aspnes [19]. We measured the real part of the sur-
face reflectance anisotropy given by the formula:
Dr
r
¼ 2
r½110Š À r½110Š
r½110Š þ r½110Š
ð1Þ
where r½110Š and r[110] are the complex reflectances along the
directions ½110Š and [110] denoted x and y, respectively.
3. Results and discussion
3.1. Quantification of the mixing domains
Despite the fact that the RAS is an optical technique, it
is very sensitive to the surface deterioration. In Fig. 1(a)
and (b), we present the STM images that have been
recorded on a freshly prepared 4° vicinal silicon surface
and on the same deteriorated silicon surface, respectively.
The corresponding RA spectra are displayed in the
Fig. 1(d) and (e) and are compared to the RA spectrum
obtained on a nominal silicon surface (Fig. 1(c)). The two
vertical lines refer to the critical points E0
0, E1 and E2,
respectively. On the vicinal surface spectrum, the total
amplitude of the anisotropy reaches 4.2 · 10À3
which is a
larger anisotropy than presented in other studies [12,13,
16,18]. The nominal silicon spectrum is similar to spectra
obtained by Shioda et al. [18,20]. The origin of the different
features observed in the spectrum corresponding to the
clean vicinal Si(001)-(2 · 1) surface, have been tentatively
interpreted by using ab initio calculations [14]. The positive
peak around 4.4 eV, located at the E2 critical point, corre-
sponds to the surface modified bulk optical transitions at
the X-point of the Brillouin zone (BZ); the small shoulder
appearing at 3.4 eV originates from the surface modified
bulk transitions at E1 and E0
0 critical points and corre-
sponds to optical transitions at L and C points in the BZ,
respectively [14,21]. This structure which is a weak shoul-
der on vicinal surfaces, appears more clearly visible on sin-
gle-domain nominal surfaces. Close to the E1 and E0
0
critical point, the structures have been shown to be sensi-
tive to the stress of the surface induced by the presence
of dimers or/and steps at the surface [21]. For lower ener-
gies, calculations show that the optical anisotropy is attrib-
uted to surface-state related transitions [14]. Around 3 eV a
broad negative structure is also observed and is more pro-
nounced on the vicinal surface spectrum than in the case of
nominal surface. This structure has been shown to be
sensitive to adsorbates and seems to be related to the
dangling bonds [13]. The interpretation of the structures
in the spectra of vicinal surface is complicated by the pres-
ence of the steps. In particular, the steps are partially
responsible for this negative minimum around 3.1 eV
[18,16,14]. The influence of the steps decreases for photon
energies larger than 3.4 eV indicating that, in this range,
the anisotropic signal mainly originates from terraces
[16]. On the nominal surface, a clear negative structure is
visible at 1.6 eV, that almost disappeared in the case of vic-
inal surface. This minimum is attributed to optical transi-
tions between occupied and unoccupied surface bands
corresponding to an hybridization along the dimer rows
of the p and pw
states of Si dimers [15,14]. It is progres-
sively reduced when increasing the miscut angle [18] and
consequently the density of steps as shown in Fig. 1(d).
The corresponding STM image Fig. 1(a) is characteristic
of the freshly prepared surface and displays very regular
terraces with the dimer rows perpendicular to the double
steps. But, we can see some places of the substrate where
the dimers are oriented in the other direction. A statistical
analysis on a 6100 nm2
surface indicates that approxi-
mately 10% of the surface is occupied with the other do-
main, in agreement with a study from Swartzentruber
et al. [22], meaning that the 4° vicinal surface is not a purely
single domain surface. Also, even on a freshly prepared
N. Witkowski et al. / Surface Science 600 (2006) 5142–5149 5143](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-82-320.jpg)
![ANNEXE1
surface, one can obtain hills occupying approximately 5%
of the surface.
After several regeneration cycles of the surface by
annealing the sample at 1050 °C for several seconds under
a pressure below 5 · 10À10
Torr, the maximum of anisot-
ropy has reduced to 2.5 · 10À3
as seen in Fig. 1(e). This sur-
face has been explored with the STM; close to well-ordered
regions we can see large hills of approximately 4–6 nm of
height which are pinning points for steps accumulation
(Fig. 1(b)). It has been already shown that annealing pro-
cedures of vicinal (100) silicon surface gives rise to the for-
mation of surface facetings [23]. The hills represent only
about 15% of the surface and can not explain the decrease
of the total anisotropy. Close to the hills, we observe sys-
tematically large areas occupied with mixing of domains.
This mixing of domains explains the decrease of the total
anisotropy in the spectrum. Now we can try to quantify
the mixing of the two domains by using the RAS technique.
In the freshly prepared surface about 5% of the surface is
covered with hills, which do not contribute to the anisot-
ropy. Consequently, the total RA signal only originates
from the other part of the surface (95%), and is therefore
smaller than the expected RA signal recorded on a defect
free surface. We can deduce that the presence of hills leads
to an undervaluation of 5% in the quantity Dr
r
. As the mea-
sured spectrum corresponds to a mixing of the two do-
mains with 10% of the dimers which are oriented
differently, we can extrapolate the RA spectrum obtained
on an ideal surface free of hills, where 100% of the dimers
are oriented in the same direction. When doing so, the RA
spectrum, called Dr
r
À Á
ideal
, has an anisotropy of 5.5 · 10À3
;
such a large anisotropy has never been observed on
Si(001) surfaces. The anisotropic signature of an actual sil-
icon surface is a linear combination of the RA signals from
the two domains and can be expressed by the expression:
A Dr
r
À Á
ideal
À ð1 À AÞ Dr
r
À Á
ideal
where A is the percentage of
the majority domain. In order to validate our hypothesis,
Fig. 2(b) shows the RA spectrum recorded on a freshly pre-
pared 2° vicinal surface (open circles) that has been in-
creased by 5% because of the presence of hills on the
surface. The simulated spectrum, in the Fig. 2(b) (plain
line), correctly reproduces the 2° vicinal surface spectrum,
except for low energies, by using the procedure described
before; the anisotropic signal Dr
r
À Á
2
is obtained by using
4nm
4nm
-3
-2
-1
0
1
Re(r/r)x10
-3
-3
-2
-1
0
1
Re(r/r)x10
-3
-3
-2
-1
0
1
Re(r/r)x10
-3
5.55.04.54.03.53.02.52.01.5
Energy (eV)
E2E1 E'0
Fig. 1. (a) STM images (current mode) 80 · 80 nm2
of the fresh 4° vicinal silicon surface; (b) STM images (current mode) 80 · 80 nm2
of the deteriorated
4° vicinal silicon surface; (c) RA spectrum of clean nominal silicon surface; (d) RA spectrum of a freshly 4° vicinal silicon surface; (e) RA spectrum of
deteriorated 4° vicinal surface.
5144 N. Witkowski et al. / Surface Science 600 (2006) 5142–5149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-83-320.jpg)
![the formula: Dr
r
À Á
2
¼ 0:76 Dr
r
À Á
ideal
À 0:24 Dr
r
À Á
ideal
. The dis-
crepancy for low energies is due to the presence of the sur-
face states that increases with the size of the terraces [18].
This rough simulation indicates us that 24% of the 2° vici-
nal surface is covered with minority domain, which is in
good agreement with results obtained by STM [22]. We
use the same procedure to extract the mixing of the two do-
mains when the 4° vicinal surface has been deteriorated by
successive annealings. The proportion of hills has been esti-
mated from STM measurements to represent approxi-
mately 13% of the surface; consequently, the RA
spectrum in the Fig. 2(a) (open circles) has been increased
by 13%. Under these conditions, the reduced anisotropy is
then only due to the mixing of the two domains. The
simulated spectrum Dr
r
À Á
4
represented in the same figure
is obtained by applying the formula Dr
r
À Á
4
¼ 0:77 Dr
r
À Á
ideal
À
0:23 Dr
r
À Á
ideal
. This clearly shows that when a 4° vicinal
surface is annealed several times, it does not remain a
single-domain surface but it presents a non-negligible
percentage of mixing domains. We demonstrated here
that the RAS is an efficient tool to characterize the sur-
face quality of vicinal silicon (001)-(2 · 1) surfaces and to
quantify the ratio of the two domains that coexist on
surfaces.
3.2. Sensitivity to adsorbates
The RA technique can also be used to study molecule
adsorption on the Si(001) surfaces. Indeed, even if the mol-
ecules do not exhibit an optical absorption when they are
adsorbed on silicon, silicon states are modified during the
adsorption and the optical transitions can be slightly af-
fected. The RA spectra that are presented in Fig. 3 have
been recorded on Si(001)-(2 · 1) after exposure to benzene
(a), ethylene (b), water (c), atomic hydrogen at 315 °C (d),
atomic hydrogen at room temperature (e), molecular oxy-
gen (f) and residual vacuum (g). In order to compare the
relative intensities of the RA spectra recorded for the differ-
ent molecules or atoms, the clean silicon spectra, which
present an anisotropic amplitude between 2.6 · 10À3
and
4 · 10À3
, have been normalized to a reference spectrum
presenting an anisotropic signal of 4 · 10À3
. The spectra,
recorded after the adsorptions, at saturation coverage,
have been normalized by using the same factor.
The benzene molecules are known to adsorb on the sur-
face in a ‘‘butterfly’’ configuration, with one dimer out of
two which is occupied [24]. The spectrum labelled (a) in
Fig. 3 is characteristic of the adsorption at saturation of
benzene on the Si(001)-(2 · 1) [25]. This spectrum is very
similar to the spectrum obtained on a partially hydroge-
nated surface (spectrum (d) plain line), where only a part
of the dangling bonds of silicon are bonded to hydrogen
atoms. When a benzene molecule is adsorbed on the
Si(001)(2 · 1) surface, it loses its aromatic character and
the optical absorption related to the p system disappears.
Accordingly, the spectrum recorded at saturation of ben-
zene reflects the modification of the silicon substrate during
the adsorption. The similarities between the spectrum
corresponding to the saturation of benzene and the spec-
trum corresponding to a partially hydrogenated surface
indicates to us that, from the optical point of view, the
hybridization between silicon and carbon or hydrogen
atom is equivalent.
In the case of ethylene, almost all the dimers are occu-
pied at saturation coverage and the molecules adsorb on-
top of the dimer with the silicon dimers remaining intact
[26]. The corresponding RA spectrum, labelled (b), is differ-
ent from the one recorded on benzene. The total anisotropy
of the spectrum is strongly reduced and positive or negative
structures are observed. One can note that all the structures
have a so-called dielectric-function-like lineshape which
has been interpreted in terms of surface local field effect
or a modification of the electron-hole interaction at the sur-
face. They do not present derivative-like lineshapes origi-
nating from a spatially or temporally localized states [27].
DFT calculations have evidenced the presence of silicon-in-
duced states and ethylene-induced states below the Fermi
level [4] that are delocalized. Transitions from silicon-in-
duced states and ethylene-induced states toward empty
states can explain the lineshapes of the structures. Espe-
cially, ethylene-induced states, denoted 1b2g, have been cal-
culated to appear between 3 and 4 eV below the Fermi level
-3x10
-3
-2
-1
0
1
Re(r/r)
5.04.03.02.0
Energy (eV)
-3x10
-3
-2
-1
0
1
Re(r/r)
Fig. 2. (a) experimental RA spectra of the deteriorated 4° vicinal silicon
surface (open circles) and calculated spectrum with 23% of the second
domain (plain line); (b) freshly prepared 2° vicinal silicon surface (open
circles) and calculated RA spectrum with 24% of the second domain (plain
line).
N. Witkowski et al. / Surface Science 600 (2006) 5142–5149 5145](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-84-320.jpg)
![ANNEXE1
and can explain the decrease of the anisotropy observed
above 4.5 eV in the RA spectrum. However, the interpreta-
tion in term of lineshape is complicated by the presence of
steps on the surface, as it will be explained in the following.
Spectrum labelled (c) has been recorded on the silicon
surfaces saturated with water whereas spectrum (d) has
been obtained when exposing the surface to atomic hydro-
gen at 315 °C. We can note that the two spectra (c) and (d)
are very similar in shape and intensity; this is not astonish-
ing since the adsorption is very similar in the two systems
as indicated on the sketches. In the case of water, the mol-
ecule is dissociated and the dangling bonds of silicon are
saturated on one side with an hydrogen atom and on the
other side with an OH specie [28], whereas in the case of
atomic hydrogen adsorbed at 315 °C, each dangling bond
is bonded to one hydrogen atom. But still some differences
are visible: below 2.5 eV, the anisotropy becomes positive
for H2O, whereas it is cancelled in the case of monohy-
dride, and the structure at E2 critical point is also more
pronounced for H2O. The interpretation of the structure
modification above 4 eV is not trivial since the origin of
this peak is not clearly established. Its origin results from
a modulation of the bulk dielectric function close to the
surface [29]. Concerning the positive anisotropy for low
energies, charge transfers from hydroxyl to silicon could
slightly modify the surface band structure leading therefore
to a non-null anisotropy.
Next, we present the spectrum recorded by exposing the
surface to atomic hydrogen at room temperature leading to
a dihydride surface (Fig. 3(e)). The spectrum shape differs
from the spectrum recorded on the monohydride spectrum,
in particular, the structures located at the E1 and E0
0 critical
points rather display a dielectric-function-like lineshape.
This observation is in contradiction to what has been de-
scribed by Mantese et al. [13]; indeed for monohydride
spectrum and dihydride spectrum, recorded on a 6° vicinal
(100) silicon surface, the RA spectra have a derivative-like
lineshape. The authors attributed these structures to an
anisotropy localized near the surface and which originates
from a dichroic bandgap due to the presence of steps on the
surface. This interpretation, utilizing steps, can be vali-
dated when comparing the monohydride spectrum re-
corded on a nominal surface which displays a clear
dielectric-function-like lineshape at the E1 and E0
0 critical
points, whereas the monohydride spectrum obtained on a
vicinal 4° surface rather displays a derivative-like lineshape
[29]. Consequently, the interpretation of the structure line-
shapes is difficult to handle in the case of vicinal surfaces.
The next example presented here concerns the adsorp-
tion of molecular oxygen at room temperature (Fig. 3(f)).
The exact mechanism of Si(001) surface oxidation at room
temperature is still under debate [30–34]. In the proposed
adsorption processes, the molecules adsorb dissociatively
and atoms incorporate progressively the Si dimers, the di-
mer back bonds and the dimer dangling bonds, leading
to a disordered surface for large coverages. RA spectrum
has been recorded for 200 L of molecular oxygen and the
corresponding RA spectrum only presents a structure at
the E1 and E0
0 critical points. In an early paper from Yas-
uda et al. [9], the authors suggest that the spectrum ob-
tained on silicon oxidized surface pertains to the
monohydride spectrum, indicating that the surface is still
dimerized. Our observation is in contradiction with this
work, since the spectrum recorded on the oxydized surface
is very close to the spectrum characteristic of dihydride
phase, where de dimers are cleaved. This similarity corrob-
orates measurements carried out using surface differential
-2x10
-3
-1
0
Re(r/r)
5.55.04.54.03.53.02.52.01.5
Energy (eV)
-2x10
-3
-1
0
Re(r/r)
-2x10
-3
-1
0
Re(r/r)
-2x10
-3
-1
0
Re(r/r)Re(r/r)Re(r/r)
E2E1 E'0
-2x10
-3
-1
0
-2x10
-3
-1
0
-2x10
-3
-1
0
Re(r/r)
a
b
c
e
f
d
g
Fig. 3. RA spectra on silicon surface saturated with: (a) benzene, (b)
ethylene, (c) water, (d) atomic hydrogen at 315 °C and partially hydro-
genated surface (plain line), (e) atomic hydrogen at room temperature, (f)
molecular oxygen, (g) clean silicon spectrum recorded after one night
under UHV; the two vertical lines indicate the position of the silicon
critical points E0
0 and E1 at 3.4 eV and E2 at 4.4 eV. The corresponding
adsorption geometry is illustrated on the side.
5146 N. Witkowski et al. / Surface Science 600 (2006) 5142–5149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-85-320.jpg)
![reflectance spectroscopy (SDRS) [34] recently published
and results based on ab initio density-functional theory
[33] where the O2 is dissociated and where oxygen atoms
are incorporated in the Si–Si dimer during initial oxidation
process. A closer inspection of first stages oxidation, using
RAS and ab initio density-functional theory, are in pro-
gress for a better understanding of the mechanisms of ini-
tial oxidation.
In the Fig. 3(g), we now present how the RA spectra are
sensitive to the modification of the silicon surface that is
contaminated with residual gases. It is well known, that
the silicon (001) surface is very reactive to any molecules,
due to the presence of dangling bonds that have an excess
or a lack of electrons [1]. A freshly prepared clean silicon
surface has been let under UHV conditions during one
night, the total pressure was 4 · 10À10
T. The residual gases
in the chamber were mainly constituted with water and
molecular hydrogen. The increase of the structure at
3.5 eV indicates us that the dangling bonds of the dimers
are progressively occupied with molecules. The spectrum
resembles that of the spectrum recorded after water
adsorption; this similarity suggests that the surface is
slowly contaminated with residual water in the chamber.
One could think the spectrum is also similar to the spec-
trum characteristic of a monohydride surface, but when
the residual gas in the chamber is almost only composed
with molecular hydrogen, which does not adsorb easily
on the silicon surface [35], the spectrum recorded after
one night is identical to the clean silicon spectrum.
3.3. Adsorption kinetics
We want to demonstrate here that RAS is also an effi-
cient tool to study the kinetics of adsorption. To illustrate
this, we present, in the Fig. 4, the RA signal recorded at
3.5 eV versus the benzene dose converted in Langmuir
(1 L = 10À6
Torr s), compared to the RA signal recorded
at 3.5 eV during atomic hydrogen adsorption at 305 °C.
The pressure of benzene during the exposure recorded with
a Bayard-Alpert type gauge calibrated to benzene, was
maintained at 1.7 · 10À7
T and the pressure of hydrogen
monitored with a mass-spectrometer, was maintained at
1 · 10À6
T. The RA signal of benzene has been extracted
from RA spectra recorded at different exposure rates [25],
whereas the RA signal of atomic hydrogen has been re-
corded during a real time measurement at 305 °C. Accord-
ing to our previous study, the evolution for lower energies
is similar to what is observed at 3.5 eV whereas the evolu-
tion at the E2 critical point follows a different profile that is
described in [25]. We have then chosen to study RA signal
recorded at 3.5 eV, because this signal is known to increase
linearly with the occupancy of the dimer dangling bonds.
When the surface is saturated with atomic hydrogen at
305 °C, a monohydride surface is obtained and almost all
the dangling bonds are occupied with a hydrogen atom,
leading to a saturation coverage close to unity [36]. We
have then normalized the hydrogen saturated signal to
unity and the same factor has been applied to benzene
curve. We can clearly see that, after saturation, the RA sig-
nal recorded on benzene saturated surface represents the
half of the signal recorded on the monohydride surface.
This corroborates the fact that RA signal is proportional
to the dimer occupancy since it is well established that ben-
zene molecules adsorb one dimer out of two, leading to a
coverage close to 0.5 [25,37]. Under these conditions,
RAS is valid to inform on the kinetics of adsorption of
atoms or molecules. We can compare here the kinetics
for two very different systems. In the case of benzene, the
molecules adsorb directly on a site without dissociation
or migration, following a Langmuirian process described
by the following equation [38]:
dh
dx
¼
½DŠ
r
ffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2pmkT
p S0eÀEa
kT ð1 À hÞ ð2Þ
where h is the coverage, x is the dose in Langmuir, [D] is a
dose correction factor, r the density of adsorption sites, S0
the initial sticking coefficient and Ea the energy of activa-
tion. When integrating the Eq. (2) one obtains a coverage
with an exponential profile as shown in the Fig. 4. Due
to inaccuracies in the pressure measurements, the first
experimental point has not been taken into account in
the calculated curve and consequently the thermodynamics
parameters can not be extracted. Similar results have been
also obtained by using Monte Carlo simulations. The situ-
ation in the adsorption of atomic hydrogen is clearly differ-
ent, since the coverage does not follow anymore an
exponential curve. For initial adsorption, the coverage in-
creases linearly, indicating that the adsorption of atomic
hydrogen obeys to a different process. As seen in the
Fig. 4, the linear increase does not start immediately after
the hydrogen introduction but is slightly delayed. This
behaviour could be due to an error in the estimation of
the initial hydrogen dose impinging the surface since the
1.0
0.8
0.6
0.4
0.2
0.0
Coverage
1.41.21.00.80.60.40.20.0
Benzene dose (L)
806040200
Hydrogen dose (L)
Fig. 4. Coverage versus benzene exposure (squares) and atomic hydrogen
exposure at 305° (crosses) together with calculated curves following a
Langmuirian process (dotted lines) or a ‘‘hot precursor’’ process (plain
line).
N. Witkowski et al. / Surface Science 600 (2006) 5142–5149 5147](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-86-320.jpg)
![ANNEXE1
mass-spectrometer, that was used to monitor the pressure,
is located far from the sample surface. To explain the
adsorption curve during hydrogen adsorption, it has been
proposed [39] that atomic hydrogen adsorption at 305 °C
follows a ‘‘hot-precursor’’ mechanism where the incoming
atoms can migrate on the surface, can visit several sites be-
fore forming pairs and finally can adsorb or desorb. This
process can be very well reproduced by using a simple
precursor-mediated adsorption model described by the
following equation [38]:
dh
dx
¼
½DŠ
r
ffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2pmkT
p S0eÀEa
kT
1 À h
1 þ hðK À 1Þ
ð3Þ
where the new parameter K is inversely proportional to the
number of visited sites. When solving the Eq. (3) by using
Euler procedure, the best curve is obtained for K = 0.18
and the agreement between the experimental and the calcu-
lated curves is excellent even for low coverages. The K
parameter is larger than the one obtained by Shioda
et al. [29] (K = 0.08) and by Widdra et al. [39] (K = 0.05).
Since this parameter increases when the number of visited
sites decreases, in our experiment, the number of visited
sites is smaller indicating that the adsorption process is
more efficient. The difference can be explained by the fact
that we did not carry out the experiment at the exact mea-
sured temperature, because the tungsten filament used to
dissociate the hydrogen molecules influences the local tem-
perature at the surface. Also, for coverages lower than 0.2,
Widdra et al. proposed that the experimental curve can
deviate from the calculated one since they expect the pair-
ing to break down due to a finite lifetime or ‘‘diffusion’’
length of the hot precursor [39]. By using the RAS tech-
nique, we have access to the very low coverages and we
can see that the calculated curve follows nicely the experi-
mental data except for the very first points for the reason
given before. Then the pairing of hydrogen atoms seems
not affecting the adsorption rate, but further investigations
are needed to better understand the processes that occur
for initial adsorption coverages.
4. Conclusion
In this article, we demonstrated the capabilities of the
RAS technique to characterize the Si(001)-(2 · 1) surfaces.
Especially, it is possible to determine the ratio of the two
perpendicular domains when knowing the roughness of
the surface. This knowledge is of particular importance
for experimentalists that study the adsorption geometry
of molecules on silicon surfaces, since two perpendicular
geometries can be observed when the surface is not a purely
single-domain one. We presented also the RA spectra for
different species of adsorbates and showed that the RA
spectra can be used as a fingerprint to discriminate between
molecules. We demonstrated, finally, that standard adsorp-
tion models can be used to reproduce the evolution of RA
spectra with the dose of adsorbate making of this technique
an efficient tool to follow the kinetics of adsorption even
for low coverages.
Acknowledgements
This work was supported by grants from Re´gion lle-de-
France.
References
[1] S.F. Bent, Surf. Sci. 500 (2002) 879.
[2] G.P. Lopinski, D.D.M. Wayner, R.A. Wolkow, Nature 406 (2002)
48.
[3] M.P. Schwartz, R.J. Hamers, Surf. Sci. 515 (2002) 515.
[4] U. Birkenheuer, U. Gutdeutsch, N. Ro¨sch, A. Fink, S. Gokhale, D.
Menzel, P. Trischberger, W. Widdra, J. Chem. Phys. 108 (1998) 9868.
[5] R.A. Wolkow, Phys. Rev. Lett. 68 (1992) 2636.
[6] P. Weightman, D.S. Martin, R.J. Cole, T. Farrell, Rep. Prog. Phys.
68 (2005) 1251.
[7] P. Chiaradia, G. Chiarotti, Photonic Probes of Surfaces, in: P. Halevi
(Ed.), Amsterdam, 1995.
[8] V.L. Berkovits, N. Witkowski, Y. Borensztein, D. Paget, Phys. Rev. B
63 (2001) 121314(R).
[9] T. Yasuda, L. Mantese, U. Rossow, D.E. Aspnes, Phys. Rev. Lett. 74
(1995) 3431.
[10] C. Noguez, C. Beitia, W. Preyss, A.I. Shkrebtii, M. Roy, Y.
Borensztein, R. Del Sole, Phys. Rev. Lett. 76 (1996) 4923.
[11] V. Mazine, Y. Borensztein, Phys. Rev. Lett. 88 (2002) 147403.
[12] L. Kipp, D.K. Biegelsen, J.E. Northrup, L.-E. Swartz, R.D. Bringans,
Phys. Rev. Lett. 76 (1996) 2810.
[13] L. Mantese, U. Rossow, D.E. Aspnes, Appl. Surf. Sci. 107 (1996) 35.
[14] W.G. Schmidt, F. Bechstedt, J. Bernholc, Phys. Rev. B 63 (2001)
045322.
[15] M. Palummo, G. Onida, R. Del Sole, B. Mendoza, Phys. Rev. B 60
(1999) 2522.
[16] S.G. Jaloviar, J.-L. Lin, F. Liu, V. Zielastek, M. McCaughan, M.G.
Lagally, Phys. Rev. Lett. 82 (1999) 791.
[17] K. Hata, T. Kimura, S. Ozawa, H. Shigekawa, J. Vac. Sci. Technol. A
18 (2000) 1933.
[18] R. Shioda, J. van der Weide, Phys. Rev. B 57 (1998) R6823.
[19] D.E. Aspnes, J.P. Harbison, A.A. Studna, L.T. Florez, J. Vac. Sci.
Technol. A 6 (1988) 1327.
[20] O. Pluchery, N. Witkowski, Y. Borensztein, Phys. Status Solidi B 242
(2005) 2696.
[21] K. Hingerl, R.E. Balderas-Navarro, A. Bonanni, P. Tichopadek,
W.G. Schmidt, Appl. Surf. Sci. 175–176 (2001) 769.
[22] B.S. Swartzentruber, N. Kitamura, M.G. Lagally, M.B. Webb, Phys.
Rev. B 47 (1993) 13432.
[23] S.M. Gray, M.K.-J. Johansson, L.S.O. Johansson, J. Vac. Sci.
Technol. B 14 (1996) 1043.
[24] N. Witkowski, F. Hennies, A. Pietzsch, S. Mattsson, A. Fo¨hlisch, W.
Wurth, M. Nagasono, M.N. Piancastelli, Phys. Rev. B 68 (2004)
115408.
[25] N. Witkowski, O. Pluchery, Y. Borensztein, Phys. Rev. B 72 (2005)
075354.
[26] F. Hennies, A. Fo¨hlisch, W. Wurth, N. Witkowski, M. Nagasono,
M.N. Piancastelli, Surf. Sci. 529 (2003) 144.
[27] U. Rossow, L. Mantese, D.E. Aspnes, Appl. Surf. Sci. 123 (1998)
237.
[28] M.K. Weldon, B.B. Stefanov, K. Raghavachari, Y.J. Chabal, Phys.
Rev. Lett. 79 (1997) 2851.
[29] R. Shioda, J. van der Weide, Appl. Surf. Sci. 130–132 (1998) 266.
[30] Yuhai Tu, J. Persoff, Phys. Rev. Lett. 89 (2002) 086102.
[31] Y. Chabal, K. Raghavachari, X. Zhang, E. Garfunkel, Phys. Rev. B
66 (2002) 161315(R).
5148 N. Witkowski et al. / Surface Science 600 (2006) 5142–5149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-87-320.jpg)
![[32] B.D. Yu, Y.J. Kim, J. Jeon, H. Kim, H.W. Yeom, I.W. Lyo, K.-J.
Kong, Y. Miyamoto, O. Sugino, T. Ohno, Phys. Rev. B 70 (2002)
033307.
[33] A. Incze, R. Del Sole, G. Onida, Phys. Rev. B 71 (2005) 035350.
[34] Y. Borensztein, O. Pluchery, N. Witkowski, Phys. Rev. Lett. 95
(2005) 117402.
[35] M. Du¨rr, Z. Hu, A. Biedermann, U. Ho¨fer, T. Heinz, Phys. Rev. B 63
(2001) 121315(R).
[36] J.J. Boland, Adv. Phys. 42 (1993) 129.
[37] Y. Taguchi, M. Fujisama, T. Takaoka, T. Okada, M. Nishijima, J.
Chem. Phys. 95 (1991) 6870.
[38] W. Ranke, Y. Joseph, Phys. Chem. Chem. Phys. 4 (2002) 2483.
[39] W. Widdra, S.I. Yi, R. Maboudian, G.A.D. Briggs, W.H. Weinberg,
Phys. Rev. Lett. 74 (1995) 2074.
N. Witkowski et al. / Surface Science 600 (2006) 5142–5149 5149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-88-320.jpg)
![ANNEXE2
Probing the Si-Si Dimer Breaking of Si 100 2 1 Surfaces upon Molecule Adsorption
by Optical Spectroscopy
Y. Borensztein,* O. Pluchery,†
and N. Witkowski‡
Institut des Nanosciences de Paris (UMR CNRS No. 7588), Universities Paris VI and Paris VII, 4 place Jussieu,
F-75252 Paris cedex 05, France
(Received 21 December 2004; revised manuscript received 20 May 2005; published 7 September 2005)
The adsorption of atoms and molecules of several gases of the Si 100 2 1 silicon reconstructed
surface is investigated by surface differential reflectance spectroscopy. This UV-visible optical spectros-
copy makes possible the discrimination between two adsorption modes, depending on whether or not the
adsorption leads to breaking the Si-Si dimers. The observation of two different optical features is assigned
to the bonding on dangling bonds or to the breaking of dimers, and gives access to the adsorption mode of
hydrogen, water, oxygen, and pyridine. Moreover, the technique being quantitative, we can determine the
total amount of dimers involved in the adsorption and monitor the adsorption kinetics.
DOI: 10.1103/PhysRevLett.95.117402 PACS numbers: 78.68.+m, 68.43.2h, 73.20.2r
In the past few years, an increasing amount of work has
been dedicated to the grafting of organic molecules on the
Si(100) surface [1]. Such hybrid systems open the way for
integrating the electronic, optical, or biological properties
of organic layers in silicon-based devices. In this so-called
bottom-up approach, the well-ordered Si 100 2 1 sur-
face can be used as a template in order to organize and
grow ordered organic layers [2]. To this end, it is critical to
understand and to control the atomic-level phenomena at
the interface between Si and the molecular layer. The
Si(100) surface, prepared in ultrahigh vacuum, displays a
2 1 reconstruction, due to the formation of rows of
parallel Si-Si dimers [3]. Each Si atom of a dimer exhibits
one dangling bond, which is particularly reactive towards
adsorption of atoms or molecules. Understanding the na-
ture of the interface between molecules and silicon re-
quires one to determine the structural changes of the Si
dimers (and their possible breaking) and of the number of
dimers involved in the bonding with the adsorbed mole-
cules [4–6]. However, the structural investigation of the
interface, by the usual laboratory surface techniques using
electrons [low-energy-electron diffraction (LEED), high-
resolution-electron-energy-loss spectroscopy] or by
scanning-tunnel microscopy (STM), is actually a difficult
task, because direct access to the interface is prevented by
the presence of the adlayer. On the contrary, techniques
based on photons can be used to study the interface through
the adsorbed thin film. Infrared spectroscopy can probe Si-
molecule bonds and internal molecule vibrations [7], but
does not inform on the Si-Si bond itself. Synchrotron-
based techniques like x-ray photoelectron diffraction
(XPD) [5], x-ray photoelectron spectroscopies [6], inelas-
tic x-ray scattering [8], and x-ray absorption spectroscopy
[9] have recently given valuable information on the inter-
face between organic molecules and Si(100). However,
these techniques require synchrotron and cannot be used
in a routine manner. Although the previous techniques
(except XPD [5]) give access to the configuration of the
adsorbed molecules, they cannot directly determine the
possible changes of the Si-Si bonds at the interface, such
as dimer breaking of the dimers. On the other hand, several
surface-sensitive linear optical techniques have been de-
veloped in the past two decades and have proved to be
efficient for studying surfaces, buried interfaces, growth of
thin films, or adsorptions [10]. They are easy-to-use and
versatile laboratory techniques. Reflectance anisotropy
spectroscopy (RAS), measuring the optical anisotropy of
the surface of a crystal [e.g., Si 100 2 1], has been
applied successfully for semiconductor [10] and metal
surfaces [11]. Surface differential reflectance spectroscopy
(SDRS) measures changes of the reflectance of a crystal,
induced by adsorption (removal of surface states, addi-
tional interface states, etc.) [10]. Moreover, real-time
SDRS, during gas exposure, reveals the adsorption kinetics
[12].
We demonstrate that SDR spectroscopy may unravel the
bonding mode of different molecules on Si 100 2 1 and
provides a clear way to discriminate between the breaking
of the surface Si dimers and their preservation upon gas
adsorption. This is shown by comparing two model cases:
the monohydride Si 100 2 1:H surface (mono-H) where
the Si dimers are intact and the dihydride Si 100 1 1:H
surface (di-H) where they are broken. The corresponding
SDR spectra are used as the ‘‘optical fingerprints’’ of the
adsorption processes. These results are compared to the
case of other adsorbed molecules (water, oxygen, pyri-
dine). SDRS giving quantitative information, we can esti-
mate the number of surface dimers involved in the bonding
and determine kinetics.
The Si 100 2 1 surfaces were prepared in ultrahigh
vacuum (5 10 11
mbar) by flashing the samples at
1050 C. Heating is achieved by direct current flow
through the samples. The samples, 4 misoriented along
the 011 direction, are constituted of 2 1-reconstructed
terraces separated by double steps, with all the dimers
parallel to the steps in the 110 direction [13]. Surface
PRL 95, 117402 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending
9 SEPTEMBER 2005
0031-9007=05=95(11)=117402(4)$23.00 117402-1 © 2005 The American Physical Society](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-89-320.jpg)
![quality was verified, not only by LEED and STM, but also
by RAS, which is very sensitive to the quality of the
reconstruction and to possible contamination. Purity of
gases was checked by use of a mass spectrometer. H2
molecules, introduced at a pressure of 10 6
mbar, were
dissociated by a hot tungsten filament located at 5 cms in
front of the sample. SDRS was performed by use of a
homemade apparatus [14], with an incidence of 45 and
with s-polarization perpendicular to the dimers.
In the first part, we consider adsorption of hydrogen
atoms (Hat) on Si 100 2 1. Adsorption at high tempera-
tures (HT) around 600 K yields a mono-H surface, where
the 2 1 organization of the surface is maintained, and
where every dangling bond (DB) is bound to a single Hat
[15]. On the contrary, adsorption at room temperature (RT)
breaks all the dimers, and leads at saturation to a slightly
disordered hydrogenated surface, with large parts of the
surface made of ordered di-hydrogenated Si atoms (as an
ideal unreconstructed Si surface saturated by H) [15]. In
this di-H phase, Si atoms are bound to two underneath Si
atoms and two Hat at the surface. These two surfaces
obtained at H saturation (mono-H at 600 K and di-H at
RT) can be considered as archetype surfaces. Figure 1
gives the corresponding SDR spectra (a) and (b), defined
by R
R
RSi RSi:H
RSi
, where RSi and RSi:H are the reflectances
of the clean and the hydrogenated surfaces, respectively. At
this point, it is important to make explicit the information
gained from SDRS. The optical reflectance RSi of the
Si 100 2 1 surface is the sum of a bulk component and
of a surface one, which originates both from the presence
of surface states and from the 2 1 symmetry of the
surface [16]. As known from density functional theory
(DFT) [17], the surface component involves numerous
electronic transitions: (i) transitions from or to specific
surface states, which can be surface-to-surface, surface-
to-bulk, or bulk-to-surface transitions; these transitions are
evidently very sensitive to any change of the surface elec-
tronic structure, for example, induced by adsorption.
(ii) Transitions between bulk electronic states in the vicin-
ity of the surface, which can be different from the same
transitions within the bulk, because of the different sym-
metry at the surface or of strain induced by the surface, and
therefore can also contribute to the ‘‘surface optical re-
sponse’’; these surface-modified bulk transitions (mainly
transitions at the so-called critical points, as indicated
below) are sensitive to more important structural changes
at the surface. When Hat is adsorbed at the surface, the
appearance of optical features in the SDR spectrum is
therefore related to the removal or to modifications of these
different transitions.
When H is adsorbed at HT, the spectrum drawn in
Fig. 1(a) displays only one main peak centered at 2.9 eV,
with a small shoulder at higher energy. At this temperature,
the surface is an almost perfect mono-H phase, where each
DB of the Si dimers is bound to 1 H atom, and the Si-Si
bond within each dimer remains intact [see inset of
Fig. 1(a)]. Consequently, the spectrum of Fig. 1(a) displays
only (or mainly) the contribution of the transitions involv-
ing the DB electronic states, and can be viewed as the
optical fingerprint of the adsorption on the dangling bonds.
It cannot be excluded that the weak shoulder around 4 eV
could be due to a small amount of hydrogen atoms incor-
porated in silicon that would break some of the Si dimers.
It is worthwhile to note that this spectrum does not display
any sharp feature at the critical point (CP) energies of
silicon, E0 E0
1 at 3.45 eV and E2 at 4.35 eV, which is
the indication that no surface-modified bulk transition is
involved in this spectrum [18]. On the contrary, when H is
adsorbed at RT, giving the di-H surface, all dimers are
broken and the 2 1 reconstruction is removed. The spec-
trum, drawn in Fig. 1(b), displays two main peaks around
3.1 and 4 eV. As the comparison is now made between the
initial clean reconstructed surface and the fully hydrogen-
ated surface with no reconstruction, whose reflectance
RSi 1 1:H is close to the one of bulk silicon [19], this
spectrum provides the intrinsic surface optical response
of the bare Si 100 2 1 surface. This experiment is, in-
deed, in good agreement with DFT calculations [13,17],
drawn in Fig. 1(c) [20]. The differences with experiment
can be due to the approximations used in the theoretical
model and to the fact that the effect of steps is not included.
In order to isolate the only contribution to SDR of the
breaking of the dimers, we have studied in more detail the
RT H adsorption, which appears to occur in a two-stage
FIG. 1. (a) SDR spectrum after complete H adsorption on
Si 100 2 1 at 600 K; (b) SDR spectrum after complete H
adsorption at 300 K; (c) DFT calculation corresponding to (b),
from Ref. [17]; (d) contribution of the dimer breaking to the SDR
signal.
PRL 95, 117402 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending
9 SEPTEMBER 2005
117402-2](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-90-320.jpg)
![ANNEXE2
process: adsorption on the dangling bonds [giving a spec-
trum similar to spectrum 1(a)], followed by the breaking of
the dimers [21]. The change from the first to the second
stage provides this contribution, and is drawn in Fig. 1(d).
This difference spectrum is therefore the optical fingerprint
of the dimer breaking, and originates both from suppres-
sion of surface transitions involving specific electronic
states of the Si-Si dimers, and from suppression of
surface-modified bulk transitions due to the removal of
the 2 1 reconstruction. This spectrum can be viewed as
a broad positive feature, extending from about 3 to 4.5 eV,
superimposed with a sharp negative one located close to
the energy 3.45 eV of the E0 E0
1 CP, which gives the
observed minimum. This minimum is therefore most likely
related to bulk transitions, which are modified in the vi-
cinity of the 2 1-recontructed surface. It has been shown,
indeed, that such features can originate from strain extend-
ing into bulk caused by surface stress related to reconstruc-
tion, dimerization, and presence of steps [18].
This approach provides a phenomenological tool which
permits us to discriminate between the adsorption on the Si
dangling bonds [Fig. 1(a)] and the breaking of the Si
dimers [Fig. 1(d)]. Spectrum 1(b) gives the sum of both
effects resulting from the complete removal of the recon-
struction and the passivation of dangling bonds.
The second part of this Letter is the validation of the
previous results and their use for investigating other ad-
sorptions. The first purpose is achieved by checking that
similar optical spectra are obtained with other adsorbed
molecules, which means that SDRS depends little on the
kind of adsorbate, but rather on the bonding of the adsor-
bate with the substrate. The case of H2O adsorbed at RT on
Si 100 2 1 is presented in Fig. 2(a). The spectrum is
very similar to the one obtained for the mono-H surface
[Fig. 1(a)], but different from the di-H one [Fig. 1(b)]. This
shows clearly that water is bound to the DBs of the dimers
and that the dimers are not broken. This is in agreement
with the dissociation picture, where water molecules are
dissociated into an Hat and an hydroxyl OH, bound to both
DBs of a dimer [22]. Moreover, the almost equal intensity
between spectra for H and H2O confirms that all Si dimers
accommodate water molecules (it is shown in the next
paragraph that SDRS gives quantitative information). The
second comparison is with the case of oxygen. Spectra (b)
and (c) of Fig. 2 have been measured after RTadsorption of
160 Langmuirs (L) and 2 L of molecular oxygen, respec-
tively. Spectrum 2(c) is very similar to spectrum 1(d), with
a smaller intensity. It means that, at small amounts, O
molecules break the dimers, removing the corresponding
internal states and reducing the surface-induced strain but
that, on the contrary, the surface states related to the
dangling bonds are preserved. The exact mechanism of
oxidation of Si(100) at RT is still under intense debate,
several mechanisms being proposed [23]. Our observation
is actually in excellent agreement with a very recently
proposed picture for the adsorption of a small amount of
oxygen (schematized in Fig. 2), where O2 is dissociated,
with one oxygen atom incorporated within the Si-Si dimer
bond leaving the DB unsaturated and the other one incor-
porated in a backbond [24]. The feature around 2.2 eV is
probably due to noise, originating from intense lines of the
deuterium lamp, although it cannot be excluded that it
could be an oxygen-related optical transition [24]. For a
larger amount, the spectrum 2(b) is similar to spectrum
1(b), which we explain as a further oxidation, which now
removes the surface states of the Si dangling bonds. The
smaller intensity with respect to the case of H [1(c)] is
likely due to incomplete oxidation of the surface at 160 L,
therefore to the preservation of Si dimers that have not
reacted.
Finally, we have also used SDRS to study the adsorption
of pyridine, which has been so far very little investigated
[25–27]. Figure 2(d) gives the spectrum obtained after
saturation at RT. It has a similar shape as the one obtained
for mono-H [Fig. 1(a)] and for water [Fig. 2(a)]. We can
conclude that pyridine molecules do not break the Si
dimers, but adsorb on Si dimers via the available dangling
bonds. Moreover, quantitative information on the amount
of adsorbed species can also be obtained from SDRS [12],
which gives an insight into the adsorption kinetics. In order
to illustrate this property, Fig. 3 shows the SDR intensity
integrated over the whole energy range, during Hat expo-
sure at 600 K. The increase is linear almost up to satura-
tion, where the Si 100 2 1:H is obtained, and does not
follow a Langmuir kinetics, whose best fitted curve is also
drawn. Such a behavior has been previously experimen-
FIG. 2. SDR spectrum after adsorption of different molecules
on Si 100 2 1 at 300 K. (a) saturation of water; (b) 160 L
oxygen; (c) 2 L oxygen ( 1:5); (d) saturation of pyridine. The
schemes on the right give the most probable configurations for
the molecule geometries on the surface.
PRL 95, 117402 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending
9 SEPTEMBER 2005
117402-3](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-91-320.jpg)
![tally obtained and is explained by the formation of ‘‘hot
precursors’’ on the surface, leading to a linear increase of
chemisorbed H [28]. This shows that SDRS gives a mea-
sure of the H coverage or, more precisely, of the number of
dangling bonds that have reacted with H. This quantitative
property of SDRS can be exploited for determining cover-
age and kinetics for other adsorptions, in particular, for
pyridine. The intensity of the SDR signal at saturation of
pyridine [Fig. 2(d)] is about 75% the one of the signal for
the mono-H phase [Fig. 1(a)]. We can therefore conclude
that about 3=4 of the dimers are affected by the adsorption.
Very recent ab initio calculations have concluded that each
pyridine molecule is adsorbed on a bridge site and is
bonded to two successive dimers on a row [26], as indi-
cated in the corresponding scheme in Fig. 2. In the ideal
case where the molecules would be perfectly ordered along
the dimer rows, all dimers would be involved in a bonding
with a pyridine molecule. Our observation that only 75% of
the dimers are affected by the adsorption is not contra-
dictory and can be explained by the proposed bridging sites
for the adsorption: the locations where molecules impinge
the surface are distributed in a stochastic way, and con-
sequently some of the dimers remain unbound (e.g., a
single dimer between two reacted pairs of dimers).
Monte Carlo calculations have shown us that, on the vici-
nal surface, the average number of dimers bound to pyri-
dine should be about 84%, which would lead to a SDR
intensity reduced to the same amount, not far from our
observation. In contrast to hydrogen, the experimental
points for pyridine, drawn in Fig. 3, are perfectly repro-
duced by a Langmuir mechanism, which is expected for
the previous adsorption picture.
In conclusion, by the use of SDRS during adsorption, we
could determine whether the Si dimers of Si(100) are
broken or not upon molecule absorption, and we could
get a good estimation of the number of dimers involved
in the bonding. Different kinetics for H and pyridine ad-
sorptions could be distinguished by real-time SDRS. This
opens the way for monitoring kinetics of adsorption, for
determining successive stages in complex adsorption pro-
cesses, and for investigating evolution of buried interfaces
between silicon and thicker organic layers.
*Electronic address: yves.borensztein@insp.jussieu.fr
†
Electronic address: olivier.pluchery@insp.jussieu.fr
‡
Electronic address: nadine.witkowski@insp.jussieu.fr
[1] J. T. Yates, Science 279, 335 (1998); M. Schulz, Nature
(London) 399, 729 (1999); G. P. Lopinski, D. D. M. Mayer,
and R. A. Wolkow, Nature (London) 406, 48 (2000); R. A.
Wolkow, Annu. Rev. Phys. Chem. 50, 413 (1999); J. M.
Buriak, Chem. Commun. 12, 1051 (1999).
[2] S. F. Bent, Surf. Sci. 500, 879 (2002).
[3] R. J. Hamers, R. M. Tromp, and J. E. Demuth, Phys.
Rev. B 34, 5343 (1986).
[4] A. J. Fisher, P. E. Blochl, and G. A. D. Briggs, Surf. Sci.
374, 298 (1997).
[5] P. Baumga¨rtel et al., New J. Phys. 1, 1 (1999).
[6] Masashi Nagao et al., Surf. Sci. 513, 413 (2002).
[7] R. J. Hamers et al., Acc. Chem. Res. 33, 617 (2000).
[8] A. Fo¨hlisch et al., Phys. Rev. B 69, 153408 (2004).
[9] N. Witkowski et al., Phys. Rev. B 68, 115408 (2003).
[10] P. Chiaradia and G. Chiarotti, in Photonic Probes of
Surfaces, edited by P. Halevi (Elsevier Science, New
York, 1995), Chap. 3; Y. Borensztein, Surf. Rev. Lett. 7,
399 (2000).
[11] Y. Borensztein et al., Phys. Rev. Lett. 71, 2334 (1993).
[12] C. Noguez et al., Phys. Rev. Lett. 76, 4923 (1996); C.
Beitia, W. Preyss, R. Del Sole, and Y. Borensztein, Phys.
Rev. B 56, R4371 (1997).
[13] Y. Borensztein and N. Witkowski, J. Phys. Condens.
Matter 16, S4301 (2004); Y. Borensztein, N. Witkowski,
and S. Royer, Phys. Status Solidi (c) 0, 2966 (2003).
[14] Y. Borensztein, T. Lopez Rios, and G. Vuye, Appl. Surf.
Sci. 41–42, 439 (1989).
[15] J. J. Boland, Surf. Sci. 261, 17 (1992).
[16] R. Del Sole, in Photonic Probes of Surfaces, edited by P.
Halevi (Elsevier Science, New York, 1995), Chap. 2;
W. G. Schmidt et al., Phys. Rev. B 67, 085307 (2003).
[17] M. Palummo et al., Phys. Rev. B 60, 2522 (1999).
[18] K. Hingerl et al., Appl. Surf. Sci. 175–176, 769 (2001).
[19] C. Hogan, R. Del Sole, and G. Onida, Phys. Rev. B 68,
035405 (2003).
[20] The intensity of the theoretical curve has been divided by
2
p
for comparison with the experiment performed at 45 .
[21] Y. Borensztein and N. Witkowski (unpublished).
[22] M. K. Weldon et al., Phys. Rev. Lett. 79, 2851 (1997).
[23] Yuhai Tu and J. Tersoff, Phys. Rev. Lett. 89, 086102
(2002); Y. J. Chabal et al., Phys. Rev. B 66, 161315
(2002); B. D. Yu et al., Phys. Rev. B 70, 033307 (2004).
[24] A. Incze, R. Del Sole, and G. Onida, Phys. Rev. B 71,
035350 (2005).
[25] Q. Li and K. T. Leung, Surf. Sci. 541, 113 (2003).
[26] H.-J. Kim and J. H. Cho, J. Chem. Phys. 120, 8222 (2004).
[27] Feng Tao et al., J. Phys. Chem. B 107, 6384 (2003).
[28] W. Widdra et al., Phys. Rev. Lett. 74, 2074 (1995).
FIG. 3. Variation of the integrated SDR signal as a function of
time during Hat exposure at T 600 K and pyrdine exposure at
room temperature. Continuous lines are best fittings within the
Langmuir model.
PRL 95, 117402 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending
9 SEPTEMBER 2005
117402-4](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-92-320.jpg)
![ANNEXE3
All-Optical determination of initial oxidation of Si(100) and its kinetics
N. Witkowski, O. Pluchery, Y. Borensztein
Institut des Nanosciences de Paris, Universit´e Pierre et Marie Curie-Paris6,
Universit Denis Diderot-Paris7, CNRS UMR 7588, 140 rue de Lourmel, F-75015 Paris, France
K. Ga´al-Nagy, A. Incze, G. Onida
European Theoretical Spectroscopy Facility (ETSF), CNISM-CNR-INFM,
and Dipartimento di Fisica dell’Universit`a di Milano, via Celoria 16, I-20133 Milano, Italy
F. Fuchs and F. Bechstedt
European Theoretical Spectroscopy Facility (ETSF),
Institut f¨ur Festk¨orpertheorie und -optik, Friedrich-Schiller-Universit¨at, Max-Wien-Platz 1, D-07743 Jena, Germany
R. Del Sole
European Theoretical Spectroscopy Facility (ETSF),
NAST and Dipartimento di Fisica dell’Universit`a
di Roma “Tor Vergata”, Via della Ricerca Scientifica, I-00133 Roma, Italy
(Dated: 7th June 2007)
By comparison of measured and ab-initio calculated surface optical spectra we demonstrate that
two main oxidation processes initially occur after dissociation of oxygen molecules, forming in both
cases Si − O − Si entities: (i) breaking of Si dimers by incorporation of oxygen atoms; (ii) incor-
poration into the silicon backbonds. The kinetics up to half-monolayer coverage is determined, and
explained in terms of Langmuir-like adsorption mechanisms with different probabilities.
PACS numbers: 78.68.+m, 73.20.At, 68.43.Mn
Numerous studies have been and are still devoted to
the initial oxidation of the Si(100) surface, which can be
considered as a paradigmatic reconstructed semiconduc-
tor surface, and which is also the industrially most rel-
evant silicon surface, e.g. in metal-oxide-semiconductor
devices, where the control of the oxide-silicon interface
is crucial. However, some important issues are not un-
derstood, in particular the microscopic mechanisms of
the oxidation, namely the adsorption sites, the surface
structural changes, and the oxygen reaction paths. Many
approaches have been used for attempting to elucidate
the mechanism of the first stages of oxidation. Scanning
Reflection Electron Microscopy combined with Auger
electron spectroscopy and core-level photoemission spec-
troscopy have shown that Si(100) oxidation proceeds
layer-by-layer [1], and that the first silicon layer is ox-
idized by molecular oxygen without energy barrier. This
has been supported by a spin-polarized first-principles
molecular-dynamics calculation [2], which showed that
the initial oxidation process is expected to proceed via
the formation of patchlike agglomerates of oxide species
distributed randomly, as has been also suggested from
experiments [3]. Several experimental techniques [4, 5],
including Scanning Tunnelling Microscopy [6] and Sur-
face Differential Reflectance Spectroscopy (SDRS) [7, 8],
and ab-initio total energy calculations [9] have also shown
that molecular oxygen dissociates and adsorb mainly on
the first silicon layer, with formation of bridge-bond Si-
O-Si entities, on the surface Si dimers and on the Si back-
bonds. However, the interplay of the two mechanisms of
incorporation and their influence on the oxidation kinet-
ics is still under debate.
Surface-sensitive optical techniques like reflectance-
anisotropy-spectroscopy (RAS) and SDRS have been
shown to be very sensitive to the geometries of recon-
structed semiconductor surfaces. They have been widely
applied to the clean Si(100) surface [10–12], which re-
constructs by dimerization of Si-Si pairs at the topmost
surface layer, forming “dimer rows” in the direction per-
pendicular to the dimer axis. The Si-Si dimers and re-
lated surface states have been recognized to be respon-
sible for most spectral features in RAS and SDRS [13].
These techniques have been successfully used to study
the adsorption of several atomic and molecular species,
in particular oxygen [7, 8, 10, 14]. Different oxidation
mechanisms are probably dominant in different temper-
ature regions, including very complex ones at high tem-
perature. However, already in the simple case of room
temperature oxidation, an understanding of the modifi-
cation of the optical response of clean Si(100) induced
by oxygen adsorption requires the use of state-of-the-art
theoretical methods and their validation by experimental
studies.
In the present Letter we combine experimental RAS
measured at room temperature for increasing expo-
sures to oxygen, and theoretical results obtained by the
presently most-reliable ab initio methods for several pos-
sible atomic geometries of the oxidized silicon surface.
This approach enables us to evidence two different mi-
croscopic processes occurring during the initial oxidation
Article soumis](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-93-320.jpg)
![2
Figure 1: (Color online) Comparison of experimental RAS
data (dashed curves) with the fitted theoretical curves (solid
line) including the structures 1D+1E+clean for different oxy-
gen exposures.
and to determine their kinetics. This demonstrates the
great potentiality of optical spectrocopies to gain infor-
mation about the atomic structure of complex interfaces.
The experiments were carried out in an ultra-high
vacuum preparation chamber with a base pressure of
5×10−11
Torr. The Si samples were highly-oriented (100)
wafers, Ph-doped with a resistivity of 0.1 to 1 Ohm·cm.
They were cleaned and reconstructed by direct continu-
ous current heating up to 1323 K. This procedure induces
electromigration of the Si atoms at the surface, and leads
to the formation of an almost single-domain (1×2) sur-
face, constituted of large majority wide (1×2) domains
separated by double-steps, and minority (2×1) domains
[10, 15]. Oxygen was introduced by use of a precision leak
valve at the pressure of 5×10−8
Torr, and the exposure
measured in Langmuir (1 L = 10−6
Torr×sec) was moni-
tored with a Bayard-Alpert-type ion gauge. The oxygen
purity was checked with a mass spectrometer. The mea-
surements were performed at room temperature, by use
of a home-made RAS apparatus, similar to the one de-
½ ¾ ½ ½
Figure 1: (Color online) Comparison of experimental RAS
data (dashed curves) with the fitted theoretical curves (solid
line) including the structures 1D+1E+clean for different oxy-
gen exposures.
and to determine their kinetics. This demonstrates the
great potentiality of optical spectrocopies to gain infor-
mation about the atomic structure of complex interfaces.
The experiments were carried out in an ultra-high
vacuum preparation chamber with a base pressure of
5×10−11
Torr. The Si samples were highly-oriented (100)
wafers, Ph-doped with a resistivity of 0.1 to 1 Ohm·cm.
They were cleaned and reconstructed by direct continu-
ous current heating up to 1323 K. This procedure induces
electromigration of the Si atoms at the surface, and leads
to the formation of an almost single-domain (1×2) sur-
face, constituted of large majority wide (1×2) domains
separated by double-steps, and minority (2×1) domains
[10, 15]. Oxygen was introduced by use of a precision leak
valve at the pressure of 5×10−8
Torr, and the exposure
measured in Langmuir (1 L = 10−6
Torr×sec) was moni-
tored with a Bayard-Alpert-type ion gauge. The oxygen
purity was checked with a mass spectrometer. The mea-
surements were performed at room temperature, by use
of a home-made RAS apparatus, similar to the one de-
½
Figure 2: (Color online) Top: top view of the optimized geom-
etry for different configuration: light spheres represent silicon
atoms, dark spheres oxygen atoms; bottom: theoretical RAS
for the corresponding oxidized structures and for clean silicon.
veloped by Aspnes et al. [16]. It delivers the relative
reflectance anisotropy of the Si(100) surface shown in
Fig. 1, given by:
∆r
r + r⊥
(1)
where r and r⊥ are the reflectances of the Si(100) sur-
face for light polarized either parallel or perpendicular to
the dimers.
Relaxed equilibrium geometries for the clean and ox-
idized surfaces (see Fig. 2 for the oxidized structures)
were computed in a standard way within DFT, using a
plane-waves basis set. Then, excited states have been cal-
culated in the independent-quasiparticle approximation,
using Kohn-Sham (KS) eigenvalues and eigenvectors as
a starting point, and neglecting both self-energy and ex-
citonic effects [17]. The computational details can be
found in Refs. [18] and [19]; in particular, KS wavefunc-
tions have been expanded up to an energy cutoff of 30 Ry.
In the calculation of the dielectric response, we apply an
upward, rigid energy shift of 0.5 eV (scissors operator)
to all the conduction bands, as the standard zeroth-order
approximation to keep into account the neglected many-
body effects. By describing the surface within a slab
geometry, one can then calculate the RAS as a function
of photon energy [20].
Figure 1 displays the experimental RAS (dashed
curves) of the Si(100) surface for increasing exposures
to oxygen. The spectrum obtained for the clean sur-
face is similar to spectra obtained previously, by using
r
= 2 Re
r − r⊥](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-94-320.jpg)
![ANNEXE3
3
the same cleaning procedure [10, 15]. It is dominated
by the typical negative-positive feature at 3.6–4.3 eV, a
negative shoulder around 3 eV and a negative peak cen-
tered at 1.6 eV, which is the signature of dimer-related
surface state transitions, from the π-like to the π*-like
states of the Si dimers, delocalized along the dimer rows.
Before discussing the adsorption of oxygen, it is relevant
to first focus on the clean surface. The corresponding
theoretical and experimental spectra are drawn in the
topmost panel of Fig. 1. It must be noticed that the-
oretical RAS are usually overestimated with respect to
experiment [13]. Consequently, the intensity of the cal-
culated spectrum has been reduced by a scaling factor
of 4.8, which is kept for the following fittings of the oxi-
dized surface RAS. The overall agreement between both
spectra is particularly satisfying, and the main features
are very well reproduced. This allows us to be quite con-
fident in the comparisons which will be presented below
for the oxidized surfaces.
When exposing the surface to oxygen up to 4.7 L, all
these features progressively decrease, in particular the
1.6 and 3.6 eV minima which are quickly smoothed out,
but a non zero signal remains overall the spectral range.
A much larger amount of oxygen (more than 100 L), is
needed in order to suppress almost completely the RA
signal [21]. As the optical response of the Si surface is
strongly sensitive to the amount of adsorbed oxygen, the
use of our model for describing the oxygen adsorption
and the optical anisotropy of the resulting surface, in
comparison with experiment, should be able to deter-
mine what happens during the adsorption at the atomic
scale. Various atomic structures involving the dissocia-
tion of one oxygen molecule have to be considered. Four
fully relaxed atomic geometries for half-monolayer (ML)
coverage, with very close adsorption energies (with differ-
ences smaller than 0.03 eV per oxygen atom), have been
obtained in previous studies [18, 19]. The most favorable
structures, labelled C1, C2, 1D and 1E, are presented in
Fig. 2. They have been optimized by total-energy mini-
mization in a (2x2) unit cell.
Structure 1D involves oxygen atoms incorporated in all
the dimers, structures 1E and C2 are similar and involve
oxygen atoms in the backbonds, but with bonds point-
ing in different directions, while C1 is a combination of
the previous structures. The RAS for each configuration
covering all the surface (corresponding to half monolayer)
are presented in Fig. 2 (lower panel), in comparison with
the RAS calculated for the clean surface. Although all
these configurations correspond to half-monolayer cov-
erage, it is striking that the RAS are very different.
When all the oxygen atoms are incorporated in the back-
bonds (1E and C2 spectra), the overall anisotropy is sup-
pressed and the spectra are almost flat without marked
features. On the contrary, when the oxygen atoms are
in the dimers, an important negative anisotropy is ob-
served with different and even larger features than for
Figure 3: (Color online) Fitting coefficients and coverage as
a function of the exposure obtained from the fitted curves
shown in Fig. 1. Coefficients of 1D, 1E reconstructions and
the coverage are well reproduced using Langmuirian processes
with the same parameter and different sticking probabilities.
the clean surface. In particular, the double feature at
3.4–4.3 eV is suppressed, while the 1.6 eV surface state
transition appears as a shoulder, shifted to 1.9 eV in an
overall negative signal. Finally, for the mixed structure
C1, the RAS is almost unchanged with respect to the
clean surface. None of these atomic structures is able to
reproduce alone the experimental RAS drawn in Fig. 1
for increasing amount of oxygen. Due to the fact that
at low coverage, several configurations with similar to-
tal energies are possible, it is likely that different geome-
tries contribute to the actual initial oxidation process. As
a consequence, it is realistic to model the Si surface at
various oxygen exposures by coexistence of clean areas
and of areas with several of the above described well-
defined low-energy structural models. Because the back-
bond models C2 and 1E both give similar quenched RAS,
comparison with experiment does not permit us to favor
one model over the other one, and we focus on the lower
energy structure, 1E. The choice of the other back-bond
structure C2 would give very similar results as the ones
presented below. Consequently, for the interpretation of
the measured RA, we consider a mixture including 1D,
1E and C1 structures only, covering fractions xD, xE and
xC of the surface (representative of contributions coming
from regions with oxygen in dimers, oxygen in backbonds
and mixed structures, respectively), together with clean
surface domains covering the fraction 1 − xD − xE − xC.
The RA can be written as a linear combination:
∆r
r
= xD ·
∆r
r
(1D) + xE ·
∆r
r
(1E) + xC ·
∆r
r
(C1)
+(1 − xD − xE − xC) ·
∆r
r
(clean) ,
(2)
where the coefficients xD, xE and xC are determined by
a least-squares fit to the experimental RAS for the dif-
ferent exposures. The first information gained from the
fit of the coefficients is that coefficient xC remains equal](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-95-320.jpg)
![4
to zero in all the coverage range: oxygen adsorb either in
configuration 1D or 1E but not in the mixed C1 configu-
ration. The resulting theoretical RA curves are drawn in
full lines in Fig. 1 and compared with experiment. The
overall agreement is good in the whole photon energy
range and for all amounts of oxygen. The progressive re-
duction of the typical features of the clean surface RAS is
well reproduced, together with the almost structureless
negative RA signal between 1.5 and 4 eV for the largest
exposure presented here.
The so-determined coefficients xD and xE (with xC =
0) represent the weights of both contributions for every
exposure. They are plotted as symbols in Fig. 3 ver-
sus oxygen exposure, together with the resulting cover-
age which is given by 0.5 · (xD + xE) ML, and with the
fraction of clean areas. The behavior of the coefficients
versus exposure is remarkably regular, showing a progres-
sive increase of the 1D and 1E weights, and of the total
coverage. From the resulting coefficients we estimate the
effective surface coverage, which reaches about 0.47 ML
for 4.7 L, in agreement with Refs. [1] and [4]. Both the 1D
contribution, related to the “oxygen-in-dimer” structure,
and the 1E one, related to the “oxygen-in-backbonds”
structure –and consequently the total coverage– appear
to follow a Langmuir-like behavior with a saturation al-
most reached around 5 L. Accordingly, a Langmuir mech-
anism for both configurations can be proposed: Incoming
O2 molecules adsorb either in the 1E or the 1D configura-
tion when impinging a clean area, with different sticking
probabilities pD and pE, with pD + pE = 1, while they
do not adsorb on an oxidized area. Within this scheme,
xD and xE fulfill the following equations as a function of
time t (linked in our experiments to the O2 exposure dose
δ by t(sec) = δ(L) · 20): dxD/dt = pD(1 − xD − xE) and
dxE/dt = pE(1 − xD − xE). The latter are solved as:
xD,E = pD,E (1 − exp(−t/τ)) = pD,E (1 − exp(−δ/δ0)).
The exposure dependence of the different coefficients can
be very well reproduced by such exponential laws, as
shown in Fig. 3 by the continuous lines. The correspond-
ing best fit parameters are: δ0 = 2.2 L, pD = 0.42, and
pE = 0.58.
In conclusion, by comparing experimental RAS results
with theoretical spectra computed for various structural
models, we developed a picture for the initial stages of
room-temperature oxygen adsorption on the Si(100) sur-
face. It is described in term of two mechanisms, (i)
incorporation of oxygen in surface dimers; and (ii) in-
corporation in backbonds. Their interplay explains the
measured RAS and its evolution with oxygen exposure,
allowing one to derive the kinetics of the two processes,
which are found to be Langmuir-like with the same expo-
nential parameter. The sticking probabilities are of the
same order of magnitude, but with a clear preference for
mechanism (ii). This all optical determination of struc-
tural and kinetical properties demonstrates the great po-
tential of optical spectroscopy, when experimental and
theoretical tools are used alltogether.
ACKNOWLEDGEMENTS
We acknowledge the European Community for finan-
cial support under the NANOQUANTA project (con-
tract no. NMP4-CT-2004-500198). Computer facilities
at CINECA granted by INFM (Project no. 352/2004 and
no. 426/2005) are gratefully acknowledged.
[1] H. Watanabe et al., Phys. Rev. Lett. 80, 345 (1998).
[2] L. C. Ciacchi and M. C. Payne, Phys. Rev. Lett. 95,
196101 (2005).
[3] Y. Hoshino et al., Surf. Sci. 488, 249 (2001); H.W. Yeom
et al., Phys. Rev. B 59, R10413 (1999).
[4] K. Nakajima, Y. Okazaki, and K. Kimura, Phys. Rev. B
63, 113314 (2001).
[5] S. Dreiner, M. Sch¨urmann, and C. Westphal, Phys. Rev.
Lett. 93, 126101 (2004); T.-W. Pi et al., Surf. Sci. 478,
L333 (2001); A. Yoshigoe and Y. Teraoka, Surf. Sci. 32,
690 (2003).
[6] H. Ikegami et al., Jpn. J. Appl. Phys. 35, 1593 (1996);
H. Itoh et al., Surf. Sci. 482, 114 (2001).
[7] Y. Borensztein, O. Pluchery, and N. Witkowski, Phys.
Rev. Lett. 95, 117402 (2005).
[8] E. G. Keim, L. Wolterbeek, and A. Van Silfhout, Surf.
Sci. 180, 565 (1987).
[9] T. Uchiyama, T. Uda, and K. Terakura, Surf. Sci. 433,
896 (1999); K. Kato and T. Uda, Phys. Rev. B 62, 15978
(2000); Y. J. Chabal et al., Phys. Rev. B 66, 161315(R)
(2002).
[10] N. Witkowski et al., Surf. Sci. 600, 5142 (2006);
O. Pluchery, N. Witkowski, and Y. Borensztein, Phys.
Status Solidi (b) 242, 2696 (2005).
[11] S. G. Jaloviar et al., Phys. Rev. Lett. 82, 791 (1999).
[12] W. G. Schmidt, F. Bechstedt, J. Bernholc, Phys. Rev. B
63, 045322 (2001).
[13] M. Palummo et al., Phys. Rev. B 60, 2522 (1999);
J.R. Power et al., Phys. Rev. B 67, 115315 (2003).
[14] N. Witkowski, O. Pluchery, and Y. Borensztein, Phys.
Rev. B 72, 075354 (2005).
[15] R. Shioda and J. van der Weide, Phys. Rev. B 57, R6823
(1998).
[16] D. E. Aspnes et al., J. Vac. Sci. Technol. A 6, 1327 (1988).
[17] G. Onida, L. Reining, and A. Rubio, Rev. Mod. Phys.
74, 605 (2002).
[18] A. Incze, R. Del Sole, and G. Onida, Phys. Rev. B 71,
035350 (2005).
[19] F. Fuchs, W. G. Schmidt, and F. Bechstedt, Phys. Rev.
B 72, 075353 (2005).
[20] R. Del Sole and R. Girlanda, Phys. Rev. B 48, 11789
(1993); F. Manghi et al., Phys. Rev. B 41, 9935 (1990);
R. Del Sole, Reflectance Spectroscopy-Theory, in Pho-
tonic Probes of Surfaces, edited by P. Halevi, (Elsevier,
Amsterdam 1995), p.131.
[21] K. Ga´al-Nagy, A. Incze, G. Onida, Y. Borensztein, N.
Witkowski, O. Pluchery, F. Fuchs, F. Bechstedt, R. Del
Sole, unpublished.](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-96-320.jpg)
![ANNEXE4
Adsorption geometry of C2H2 on the single-domain Si(0 0 1)-
(2 · 1) surface: fully polarization resolved NEXAFS
Annette Pietzsch a
, Franz Hennies a
, Alexander F€ohlisch a,*, Wilfried Wurth a
,
Mitsuru Nagasono a,b
, Nadine Witkowski c
, Maria Novella Piancastelli d
a
Institut f€ur Experimentalphysik, Universit€at Hamburg, Luruper Chaussee 149, D-22761 Hamburg, Germany
b
Department of Material Science and Engineering, Graduate School of Engineering, Kyoto University, Sakyo-ku 606-8501, Kyoto, Japan
c
Laboratoire d’Optique des Solides, UMR 7601, Universite Pierre et Marie Curie, 4 Place Jussieu, F-75252 Paris Cedex 05, France
d
Department of Chemical Sciences and Technologies and INFM, University of Roma ‘‘Tor Vergata’’, I-00133 Roma, Italy
Available online 31 May 2004
Abstract
The adsorption of acetylene on single-domain Si(0 0 1)-(2 · 1) surfaces has been investigated at room temperature
using fully polarization resolved near edge X-ray absorption fine structure (NEXAFS) spectroscopy. Two coexisting
adsorption species have been observed, which we ascribe to the dimerized and end-bridge structures. In both cases, the
Si dimers remain intact and the acetylene carbon atoms rehybridize to sp2
.
Ó 2004 Elsevier B.V. All rights reserved.
Keywords: X-ray absorption spectroscopy; Chemisorption; Silicon; Alkynes; Near edge extended X-ray absorption fine structure
(NEXAFS)
1. Introduction
As the demand for SiC containing devices has
increased in optoelectronic applications, the
understanding of hydrocarbon adsorption and
bonding on semiconductors is of great interest.
Especially acetylene (C2H2) adsorption has been
the subject of investigation [1–12]. The precise
adsorption geometry of acetylene is still under
debate, since calculations show a multitude of
possible structures; some being energetically very
close to each other.
In an early study, Nishijima et al. [1] investi-
gated acetylene adsorption on silicon using high
resolution electron energy loss spectroscopy
(HREELS) and low energy electron diffraction
(LEED). It was determined that acetylene pre-
dominantly chemisorbs non-dissociatively on the
Si(0 0 1) dimerized surface in the temperature
range 80–300 K. The acetylene molecule was found
to adsorb on top of a Si dimer bound to two
adjacent silicon atoms (dimerized, see Fig. 1(a),
saturating the dangling bonds and leaving the Si
dimer intact. A few years later, in a study of acet-
ylene adsorption using Auger electron spectros-
copy, temperature-programmed desorption (TPD),
and HREELS [3,4], it was proposed that the Si
dimer is cleaved by adsorption on top of the dimer
(broken-dimer structure, as shown in Fig. 1(b).
*
Corresponding author. Tel.: +49-40-8998-3122; fax: +49-
40-8998-2179.
E-mail address: alexander.foehlisch@desy.de (A. F€ohlisch).
0039-6028/$ - see front matter Ó 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.susc.2004.05.007
Surface Science 562 (2004) 65–72
www.elsevier.com/locate/susc](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-97-320.jpg)
![Early classical trajectory and slab calculations [6,5]
also favored the broken-dimer structure, whereas
subsequent density functional theory, first princi-
ples dynamics, and total energy surface studies [7–
9] found the dimerized structure with an intact Si
dimer to be considerably more stable.
The dimerized configuration as the groundstate
of chemisorption was also proposed by Hofer et al.
[14] in simulating scanning tunneling microscopy
(STM) images and calculating adsorption energies.
Apart from the broken-dimer structure and the
dimerized structure with an adsorption energy of
2.97 eV, three alternative adsorption structures
were considered: The end-bridge structure (Fig.
1(c), with the CAC bond bridging the ends of two
dimers and having 2.87 eV adsorption energy.
There, the carbon atoms are sp2
hybridized as
supported by HREELS [15] (like in the dimerized
and broken-dimer structures), thus the acetylene
contains a CAC double bond. Apart from that,
two 4-fold bonded configurations (tetra-r bonds)
have been proposed by Xu et al. [10] based on
photoelectron diffraction measurements. Here, the
acetylene retains only a single CAC bond and is
thus sp3
hybridized. Two orientations were con-
sidered: The acetylene molecule bonds to two
adjacent Si-dimers with the CAC bond parallel to
the dimer axis (p-bridge structure, Fig. 1(d), or the
CAC bond axis is oriented perpendicular with re-
spect to the dimer axis (r-bridge structure, Fig. 1(e).
The distance between the C atoms in sp3
hybrid-
ization is increased by about 0.2 A with respect to
the sp2
hybridization. The adsorption energies of
the r-bridge and p-bridge species are found to be
2.00 and 1.20 eV, respectively [14].
It has also been considered that different
adsorption species coexist on the surface: Using
scanned-energy photoelectron diffraction, Terborg
et al. [16] found evidence for the coexistence of at
least two distinct species, which they determine to
be the dimerized species and a tetra-r configura-
tion. By comparing calculated HREEL spectra
with experimentally reported spectra, Morikawa
[17] stated that at least two energetically very close
stable adsorption states coexist: At a coverage of
0.5 ML, the dimerized structure is predicted to be
the most stable configuration, while at 1 ML
coverage in turn the end-bridge structure becomes
the most stable configuration (that means a pair of
end-bridge configurations occupying the same two
Si-dimers), in agreement with the work of Sorescu
and Jordan [19]. This configuration was also iden-
tified by Kim et al. [20] using STM and pseudo-
potential calculations. Recent first-principle
calculations [21,22] also favor the coexistence of
the dimerized and end-bridge structures. Silvestr-
Fig. 1. Acetylene adsorption models on Si(0 0 1)-(2 · 1): (a) di-
merized model with C2v symmetry [1], (b) broken-dimer model
with C2v symmetry [3], (c) end-bridge model with Cs symmetry
[10], (d) p-bridge model with C2v symmetry, and (e) r-bridge
model with C2v symmetry; from [13]. At the bottom, the used
coordinate system relative to the acetylene molecules is shown.
(For a colour version of the figure see the online paper.)
66 A. Pietzsch et al. / Surface Science 562 (2004) 65–72](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-98-320.jpg)
![ANNEXE4
elli et al. [21] also found the lowest-energy end-
bridge structure to be stable and non-metallic in
contradiction to Miotto et al. [23].
In contrast, Yeom et al. [24] did not find any
evidence for a coexistence of the end-bridge and
dimerized species investigating the acetylene
adsorption using core-level photoemission spec-
troscopy. They explained the discrepancy by the
fact that coexistence may happen only for the very
initial adsorption or that the C 1s binding-energy
differences are too small to be resolved. However,
they admit that the end-bridge structure is chemi-
cally very similar to the dimerized bonding and
thus might not be distinguished.
NEXAFS on C2H2/Si(001)-(2 · 1) has been
measured earlier by Matsui et al. [11,12]. There,
the adsorption of acetylene has been studied for
different orientations of the polarization vector
with respect to the surface normal. However, the
spectra were not taken on samples with oriented
dimers. Thus, the obtained information about the
adsorption geometry is limited.
A complication in the study of the adsorption
geometry is that the Si(0 0 1) surface exhibits a
mixture of 90° rotated domains of Si dimers. To
investigate the orientation of adsorbed C2H2, we
need single domain Si(0 0 1) surfaces in order to
determine the spatial orientation of the adsorbed
molecule relative to the dimer reconstructed sur-
face. A single domain Si(0 0 1)-(2 · 1) surface has
been successfully prepared by using a Si(0 0 1)
crystal miscut by 5°. For a Si crystal cut directly in
(0 0 1), one atomic layer steps will occur at the
surface and the formed silicon dimers will be ro-
tated by 90° at every 1-layer step [25]. In the miscut
crystal however, only 2-layer steps occur, leading
to an uniform orientation of the dimer rows [25].
On vicinal cut Si(0 0 1)-(2 · 1) single domain
surfaces, we have successfully used carbon K-edge
NEXAFS to determine the unoccupied electronic
structure and geometric orientation of adsorbed
C2H4 [18] and C6H6 [26] on the Si(0 0 1)-(2 · 1)
surface.
In the present paper, we investigate the bonding
of C2H2 at saturation coverage on the Si(0 0 1)-
(2 · 1) single-domain surface, using polarization
dependent NEXAFS. This allows us to observe the
unoccupied valence states of this adsorbate system
and to determine the spatial orientation of the
adsorbed acetylene relative to the Si(0 0 1) surface,
going beyond NEXAFS using unoriented domains
[11,12], where the orientation of the adsorbate in
the surface plane could not be distinguished. We
interpret our NEXAFS data as due to the coexis-
tence of two adsorption species, which we ascribe
to the dimerized and end-bridge structures in a
ratio of 5:4.
2. Experiment
The experiments have been carried out at the
surface endstation on the undulator beamline I511
at the Swedish national synchrotron facility MAX-
Lab in Lund which provides linear polarized
radiation in an energy range of 100–1500 eV [27].
The end station consists of two connected UHV
chambers: a preparation chamber operated at low
10À10
Torr, and an analysis chamber where X-ray
photoemission spectroscopy (XPS), NEXAFS,
and X-ray emission spectroscopy (XES) experi-
ments can be performed in the 10À11
Torr range.
The analysis chamber is rotatable around the
optical axis of the incoming beam. This allows to
freely vary the relative angle between the electric
field vector of the synchrotron radiation, the
sample, and the angle of detection.
NEXAFS was carried out in constant final state
mode detecting carbon KLL Auger decay in an
energy interval of 228–278 eV with a Scienta
SES200 electron analyzer. The bandwidth of the
incoming photons was set to 50 meV and the
photon energy was determined to an accuracy of
0.1 eV using the first- and second-order of the
beamline grating monochromator. The NEXAFS
spectra were doubly normalized [28]: Each spec-
trum was normalized to the incident photon flux,
measured on a reference gold mesh. Additionally,
spectra of the clean substrate were subtracted from
spectra of the adsorbate covered surface. Finally,
all spectra have been normalized to unit height at
the continuum region at hm ¼ 315 eV.
The molecules were adsorbed on 5° vicinal cut
single domain Si(0 0 1)-(2 · 1). We mounted two
1 · 1 cm2
single domain Si(0 0 1)-(2 · 1) crystals
taken from the same 5° vicinal cut Si(0 0 1) wafer
A. Pietzsch et al. / Surface Science 562 (2004) 65–72 67](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-99-320.jpg)
![at 7° incidence angle on a tantalum plate with
molybdenum clamps. The surface structure and
the orientation was checked with LEED. For one
Si crystal the [1 1 0] direction is along the incident
photon beam, whereas for the other one the [1 1 0]
direction is along the incident photon beam.
With this setup, the electric field vector of the
incident radiation can be oriented normal to the
surface, and in the surface plane with the electric
field vector either normal or parallel to the Si di-
mers.
The samples were outgassed at 800 K for about
12 h under UHV conditions. To clean the sample,
several cycles of sputtering with Xeþ
ions at room
temperature were applied, followed by subsequent
annealing to 1250 K to reconstruct the surface.
The sample cleanliness was checked using XPS.
Saturation coverage of acetylene gas was dosed at
room temperature.
3. Results and discussion
In Fig. 2, carbon K-edge NEXAFS of C2H2 on
Si(0 0 1)-(2 · 1) for out-of-plane polarization ( n!)
for the two samples are presented. As shown in the
schematic drawing inset, the electric field vector is
normal to the Si dimers for both samples. In both
cases, a similar spectral distribution is observed
which further emphasizes the equivalence of the
two Si(0 0 1) crystals. In particular, two sharp
features at 283.8 and 286.7 eV and a broader
structure at 288.4 eV have been observed.
Turning the electric field vector of the incident
radiation into the surface plane, we obtain the
NEXAFS spectra shown in Fig. 3. With the elec-
tric field vector along the Si dimers (sk polariza-
tion) we observe the three states at 283.8, 286.7,
and 288.4 eV as for n! polarization plus an addi-
tional broad feature at 299 eV. With the electric
field vector perpendicular to the Si dimers (s?
polarization) we also observe these four peaks.
The first resonance at 283.8 eV has about 20%
higher intensity with respect to the continuum step
height at 315 eV for s? polarization.
The resonances at 283.8 and 286.7 eV show a
strong variation of the peak intensities with
polarization. The first resonance is dominating the
spectra with s? and sk polarization, whereas it only
contributes to a very small amount to the n!
spectra. On the other hand, the resonances at 286.7
and 288.4 eV are observed to increase for n!
polarization. The intensity of the second resonance
at 286.7 eV is found to be about one and a half
times as large in the n! as in the s! geometries.
Since the resonance at 283.8 eV is most pro-
nounced in the s? and sjj geometry, in-plane spatial
orientation of this state is suggested. The reso-
nance state at 286.7 eV, showing its maximum
intensity for out-of-plane polarization, is thus
stated to be oriented perpendicular to the surface.
A similar behavior is observed for the peak at
288.4 eV.
For the following discussion, a coordinate sys-
tem relative to the acetylene molecules is used; it is
Photon Energy (eV)
Intensity(arb.units)
n
n
310300290280
310300290280
C-Si
Fig. 2. NEXAFS spectra of C2H2 on Si(0 0 1)-(2 · 1) for ~E
vector normal to the surface.
68 A. Pietzsch et al. / Surface Science 562 (2004) 65–72](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-100-320.jpg)
![ANNEXE4
shown in the lower part of Fig. 1. Now, based on
the polarization-dependence, we can directly as-
sign the two observed sharp resonances at 283.8
and 286.7 eV to states which derive from the C 2p-
type pÃ
CAC states perpendicular to the molecular
axis which represent the antibonding part of the
triple bond in the free acetylene molecule. The
state at 283.8 eV is observed with very high
intensity in the s? and sjj geometries and with
much lower intensities also in the n! geometry. The
intensity ratio is about 6:1 for s? : n! and 5:1 for
sk : n!. This confirms the orientation of this state
parallel to the surface; it is thus assigned to a py-
derived pÃ
CAC state. The orientation of this pÃ
CAC
state implies the orientation of the CAC axis of the
adsorbed molecules parallel to the surface.
Since the state at 286.7 eV is observed to be
perpendicular to the surface, it is stated to be de-
rived from the pz states. Its orientation suggests
that it is involved in the surface bond between the
acetylene molecule and the substrate. Matsui et al.
[12] assign the second resonance at 286.7 eV to a
state which they denote as rÃ
C–Si. From the ob-
served polarization dependence, this resonance is
denoted to be a CASi resonance. Comparing this
with ethylene adsorption on Si(0 0 1)-(2 · 1) [18],
the CASi state is also observed to be most prom-
inent in the corresponding measuring geometry
[12]. However, its energy is lower by 1.4 eV due to
the weaker bonding.
As the free acetylene has a sp-hybridization, it
will rehybridize to sp2
or even sp3
during bonding.
The resulting hybridization depends on the struc-
ture of bonding: sp3
-hybridization allows bonding
to four Si atoms, whereas sp2
-hybridization only
leaves space for two new bonding partners. The
sharp resonance peak at 283.8 eV representing an
undisturbed pÃ
CAC state is observed in every mea-
surement geometry, thus a tetra-r bond seems
unlikely. The observation of two resonances
resulting from the splitting of the pÃ
resonance of
the free acetylene molecule further supports this
statement. This leaves the dimerized, broken-
dimer and end-bridge structures for the adsorption
species. From the intensity variation of the CASi
resonance, it may be concluded that this bond is
oriented mainly perpendicular to the surface. Its
contribution to the spectra is highest for n!
polarization and decreases towards s! polarization
which coincides well with the dimerized and end-
bridge structures. For the broken-dimer structure
however, a CASi bond with nearly 45° angle to-
wards the surface is expected due to the larger
SiASi distance. Thus, this adsorption geometry
can also be excluded.
As a consequence of the adsorption, also the
CAH bond does not longer lie along the molecular
axis. As the p-type unoccupied CAC orbital splits
up, it not only forms the CASi bond, but part of it
joins the CAH bond and causes it to bend upwards
away from the surface, as has also been observed
for ethylene adsorption on Si(0 0 1)-(2 · 1) [18].
The feature at 288.4 eV shows exactly that
behavior. Its intensity is strongest for excitation
Photon Energy (eV)
Intensity(arb.units)
s
s
310300290280
310300290280
C-Si
Fig. 3. NEXAFS spectra of C2H2 on Si(0 0 1)-(2 · 1) for ~E
vector parallel to the surface.
A. Pietzsch et al. / Surface Science 562 (2004) 65–72 69](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-101-320.jpg)
![with n!-polarization and it is in good agreement
with the data from [12]. Thus, this peak is assigned
to be a rÃ
CAH state.
As noted above, the intensity of the first reso-
nance is nearly equally large for both sjj- and s?-
polarization excitation, see Fig. 3. This indicates
that the acetylene molecules do not adsorb on the
surface in a defined azimuthal orientation, but are
either randomly oriented or adsorb in at least two
different structures which are rotated with respect
to each other. Measurements of polarization re-
solved NEXAFS of ethylene on Si(0 0 1)-(2 · 1)
performed by us [18] using the same wafer resulted
in a clear difference between the sjj and s? spectra,
thus in contrast showing a single adsorption
geometry with C2 symmetry. Hence, a possible
scenario would be the coexistence of the dimerized
and the end-bridge structure (as proposed in
[17,20]), see Fig. 1. Alternative models describing a
random orientation of the adsorbed molecules are
not supported by theoretical investigations. An
adsorption species oriented at 45° with respect to
the Si dimers has been considered by Sorescu and
Jordan [19] and has been found to be very unsta-
ble.
Turning to the continuum resonances, we ob-
serve a very broad feature at 299 eV. It is visible
in both s? and sk polarization and weak in n!.
The energy position of the peak at 299 eV is in
quite good coincidence with the rÃ
C–C state in [12]
where dimerized adsorption is stated. As this state
is oriented along the CAC bond, its observation
allows deductions about the orientation of these
bonds: The peaks at 299 eV are most intense for
s! polarization, thus the CAC bond is oriented
parallel to the surface further supporting our
conclusions based on the behavior of the pÃ
reso-
nance. The position of this resonance also allows
some conclusions about the CAC bond length
and the hybridization. Comparing the NEXAFS
spectra for gas phase C2H2 (sp-hybridized, CBC
triple bond), C2H4 (sp2
-hybridized, C@C double
bond), and C2H6 (sp3
-hybridized, CAC single
bond), one notices a shift of the rÃ
-resonance to
lower energy [28]. In the case of C2H4, the rÃ
-
resonance is shifted from about 300 eV for the
free molecule to 291 eV for the adsorbed molecule
[18]. This could be explained by the rehybridiza-
tion of the C@C double bond of the free C2H4
molecule to a single bond of the adsorbate com-
parable to that of free C2H6, but still being
somewhat shorter. In the case of C2H2, the rÃ
-
resonance is thus expected at about 300 eV for a
ethylene-type sp2
adsorption (i.e. dimerized, end-
bridge) and at about 291 eV for a ethane-type sp3
adsorption (i.e. r-bridge, p-bridge). Since in both
spectra a rCAC resonance at 299 eV is observed,
the acetylene hybridization is stated to be sp2
for
both adsorption species. The remaining pÃ
CAC
resonance confirms the existence of a C@C double
bond. The sp3
hybridization would lead to a tet-
rahedrical state which would also be observed in
the n! spectra. Thus it is concluded that the
adsorption species aligned parallel to the Si di-
mers is a dimerized species. This structure has
been calculated to be the most stable adsorption
structure together with the end-bridge configura-
tion [19,21,29]. The sp2
hybridization of the CAC
bond has also been observed using HREELS
which identified the hybridization based on the
C@C stretching frequency [15]. The latter being
slightly lower for acetylene in gasphase or on
metal surfaces, it indicates a weakened but still
remaining C@C bond after adsorption.
Also the rotated (end-bridge) adsorption species
contains a C@C double bond which should show
in the sk-excited spectrum as a pronounced pÃ
CAC
peak at 283.8 eV as is indeed observed. Investi-
gating the different adsorption geometries, it is
evident that the only structure aligned perpendic-
ular to the Si dimers and leaving the observed pCAC
bond undisturbed is the end-bridge configuration.
The CAC bond length for that geometry has been
calculated to be about the same value as for the
dimerized case (1.37 and 1.36 A, respectively) [29].
Thus, the CAC bonds of both adsorption species
contribute to the rÃ
resonance at 299 eV; this is
supported by the fact that the resonance is not
visible in the n!-excited spectra, but observed for
both sjj and s? polarized excitation.
We can now estimate the relative coverage of
the dimerized and the end-bridge adsorption spe-
cies based on the relative peak intensity of the
283.8 eV pÃ
CAC resonance in the s?- and sk-excited
spectra. Since the corresponding orbital is oriented
perpendicular to the acetylene molecular axis, the
70 A. Pietzsch et al. / Surface Science 562 (2004) 65–72](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-102-320.jpg)
![ANNEXE4
s? spectrum shows the contribution from the di-
merized adsorbed molecules. The end-bridge ad-
sorbed molecules then contribute to the sk
spectrum. The pÃ
CAC resonance is about 20% higher
for s? polarization than for sk polarization.
Assuming for both species the same transition
strength, the ratio of dimerized to end-bridge
species is 5:4 for measurement at room tempera-
ture. Changing the temperature will presumably
shift the saturation coverage and equilibrium
population of the two adsorption species.
Until now, the NEXAFS spectra are discussed
assuming that all features arise from acetylene
molecules adsorbed on the terraces. In principle,
there might be some contribution from molecules
situated at the step edges. In order to investigate
this contribution we have taken spectra with a
polarization angle of ±45° towards the surface
shown in Fig. 4. These spectra should show a clear
difference if step species were important. However,
this is not the case. Here we conclude that the
spectral contribution from molecules adsorbed on
the step edges has to be small.
4. Conclusion
In conclusion, acetylene adsorbs at room tem-
perature and saturation coverage in more than one
bonding configuration on the Si(0 0 1)-(2 · 1) sur-
face where the carbon atoms are sp2
hybridized
and the CAC bond lies in the surface plane. This
we interpret as due to a mixture of dimerized and
end-bridge species in a ratio of 5:4, where the CAC
axis is oriented in the surface plane parallel and
perpendicular to the Si-dimers, respectively. Thus,
the investigation of acetylene adsorption on single
domain Si surfaces fully resolving the polarization
dependence allows unambiguous determination of
the adsorption geometries and gives clear evidence
for the simultaneous existence of more than one
adsorbate species.
Acknowledgements
We gratefully acknowledge help from Lisbeth
Kjeldgaard and the MAX-Lab staff. This work
Intensity(arb.units)
310300290280
Photon Energy (eV)
310300290280
Photon Energy (eV)
Fig. 4. NEXAFS spectra of C2H2 on Si(0 0 1)-(2 · 1) for the incident beam in [1 1 0] (left) and [1 1 0] (right) and polarization at +45°
and )45° towards the surface.
A. Pietzsch et al. / Surface Science 562 (2004) 65–72 71](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-103-320.jpg)
![was supported by the DFG grant Fo 343/1-1 and
the Grant-in-Aid for Japan Society for the pro-
motion of Science Fellows.
References
[1] M. Nishijima, J. Yoshinobu, H. Tsuda, M. Onchi, Surf.
Sci. 192 (1987) 383.
[2] L. Li, C. Tindall, O. Takaoka, Y. Hasegawa, T. Sakurai,
Phys. Rev. B 56 (8) (1997) 4648.
[3] P.A. Taylor, R.M. Wallace, C.C. Cheng, W.H. Weinberg,
M.J. Dresser, W.J. Choyke, J.T. Yates Jr., J. Am. Chem.
Soc. 114 (1992) 6754.
[4] C. Huang, W. Widdra, X.S. Wang, W.H. Weinberg, J. Vac.
Sci. Technol. A 11 (4) (1993) 2250.
[5] B.I. Craig, P.V. Smith, Surf. Sci. 276 (1992) 174.
[6] C.S. Carmer, B. Weiner, M. Frenklach, J. Chem. Phys. 99
(1993) 1356.
[7] Q. Liu, R. Hoffmann, J. Am. Chem. Soc. 117 (1995) 4082.
[8] Y. Imamura, Y. Morikawa, T. Yamasaki, H. Nakasuji,
Surf. Sci. 341 (1995) L1091.
[9] R.H. Zhou, P.L. Cao, L.Q. Lee, Phys. Rev. B 47 (16)
(1993) 10601.
[10] S.H. Xu, M. Keefe, Y. Yang, C. Chen, M. Yu, G.J.
Lapeyre, E. Rotenberg, J. Denlinger, J.T. Yates, Phys.
Rev. Lett. 84 (5) (2000) 939.
[11] F. Matsui, H.W. Yeom, A. Imanishi, K. Isawa, I.
Matsuda, T. Ohta, Surf. Sci. 401 (1998) L413.
[12] F. Matsui, H.W. Yeom, I. Matsuda, T. Ohta, Phys. Rev. B
62 (8) (2000) 5036.
[13] A. Fink, Ph.D. thesis, Technische Universit€at M€unchen,
2001.
[14] W.A. Hofer, A.J. Fisher, R.A. Wolkow, Surf. Sci. 475
(2001) 83.
[15] W. Widdra, C. Huang, S.I. Yi, W.H. Weinberg, J. Chem.
Phys. 105 (3) (1996) 5605.
[16] R. Terborg, M. Polcik, J.T. Hoeft, M. Kittel, D.I. Sayago,
R.L. Toomes, D.P. Woodruff, Phys. Rev. B 66 (2002)
085333.
[17] Y. Morikawa, Phys. Rev. B 63 (2001) 033405.
[18] F. Hennies, A. F€ohlisch, W. Wurth, N. Witkowski,
M. Nagasono, M.N. Piancastelli, Surf. Sci. 529 (2003)
144.
[19] D.C. Sorescu, K.D. Jordan, J. Phys. Chem. B 104 (2000)
8259.
[20] W. Kim, H. Kim, G. Lee, Y.K. Hong, K. Lee, C. Hwang,
D.H. Kim, J.Y. Koo, Phys. Rev. B 64 (2001) 193313.
[21] P.L. Silvestrelli, O. Pulci, M. Palummo, R. Del Sole, F.
Ancilotto, Phys. Rev. B 68 (2003) 235306.
[22] J.H. Cho, L. Kleinman, Phys. Rev. B 69 (2004) 075303.
[23] R. Miotto, A.C. Ferraz, G.P. Srivastava, Phys. Rev. B 65
(2002) 075401.
[24] H.W. Yeom, S.Y. Baek, J.W. Kim, H.S. Lee, H. Koh,
Phys. Rev. B 66 (2002) 115308.
[25] S. van Dijken, H.J.W. Zandvliet, B. Poelselma, Surf. Rev.
Lett. 5 (1998) 15.
[26] N. Witkowski, F. Hennies, A. Pietzsch, S. Mattsson, A.
F€ohlisch, W. Wurth, M. Nagasono, M.N. Piancastelli,
Phys. Rev. B 68 (2003) 115408.
[27] R. Denecke, P. V€aterlein, M. B€assler, N. Wassdahl, A.
Nilsson, J.-E. Rubensson, J. Nordgren, N. Martensson, R.
Nyholm, J. El. Spec. Rel. Phen. 101–103 (1999) 971.
[28] J. St€ohr, NEXAFS Spectroscopy, in: Springer Series in
Surface Science, vol. 25, Springer-Verlag, Berlin, 1992.
[29] P.L. Silvestrelli, F. Toigo, F. Ancilotto, J. Chem. Phys. 114
(19) (2001) 8539.
72 A. Pietzsch et al. / Surface Science 562 (2004) 65–72](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-104-320.jpg)

![2
and co-workers12
at 1 ML coverage the bridge geometry
is energetically favorable (see Fig.1 for a schematic
representation of the different geometries of adsorption).
No experimental observation has evidenced so far the
presence of the bridge geometry, and this contradiction
between experimental observations and theoretical
results could stem from the kinetics of adsorption, as
pointed out in13
. The coverage during the adsorption
process and after saturation is an important issue for
determining the adsorption configuration. STM images2
revealed that the molecule-molecule interaction is not
negligible during the adsorption process. The images
showed that ethylene molecules tend not to stay on top
of neighboring dimers. As a consequence, it was argued
that 0.5 ML (i.e. one molecule for every second dimer)
could be the saturation coverage. ARPES experiment5
,
instead, showed that, at saturation coverage, only the
adsorption of one molecule per dimer could explain the
periodicity of the observed spectra in the reciprocal
space. A recent core-level spectroscopy experiment15
found a saturation coverage of 0.87 ML. This result
is confirmed by Surface Differential Reflectance Spec-
troscopy (SDRS) measurements16
where a coverage of
0.90 ± 0.05 ML is found at saturation, following the
same procedure as in Refs.18,19. Moreover it appears
from previous experimental studies20
and from ours16
that the sticking coefficient decreases dramatically
beyond 0.5 ML coverage. Consequently, a large amount
of ethylene exposure is needed in order to reach a full
monolayer coverage. This fact could explain the differ-
ent interpretations concerning the coverage at saturation.
In this paper, we present a combined experimental
and theoretical investigation, which permits us to con-
firm most of the previous statements and to solve several
unclear issues, in particular the discrepancy between
experiment and theory concerning the adsorption config-
uration at 1 ML coverage. Indeed, from a pure energetic
point of view, the bridge configuration is found to be
the favored one for 1 ML. The most important point
of this paper is that the comparison between the ex-
perimental spectra measured by Reflectance Anisotropy
Spectroscopy (RAS)17
and the calculated ones, clearly
demonstrates that from 0.5 ML to 1 ML coverage, the
adsorption geometry is the on-top one. This discrepancy,
between what is expected and what is observed, can be
explained by kinetic effects which prohibit the evolution
from the on-top configuration to the bridge one, when
increasing the coverage above 0.5 ML. Our results about
the atomic structure confirm that silicon dimers do not
break during ethylene adsorption.
II. EXPERIMENTAL AND COMPUTATIONAL
DETAILS
A. Experimental details
The experiments were carried out in an ultrahigh vac-
uum (UHV) preparation chamber with a base pressure of
5×10−11
Torr, equipped with in situ low-electron energy
diffraction, RAS and SRDS apparatus. The Si(001) sam-
ples were vicinal surfaces with a 4◦
miscut towards the
[110] direction, in order to have single-domain surfaces
constituted of terraces of about 3-4 nm with the dimer
rows aligned along the [110] direction. Preparation of
the surface is described in details in Ref.21. Ethylene
was introduced by use of a precision leak valve, purity of
the gas was checked with a mass spectrometer and the
exposure was monitored with a Bayard- Alpert-type ion
gauge.
All the optical measurements were performed at room
temperature, by use of a home-made RAS apparatus
similar to the one developed by Aspnes et al.22
. The
anisotropy of the surface reflectance is given by the for-
mula :
∆r
r
= 2
r[¯110] − r[110]
r[¯110] + r[110]
(1)
where r[¯110] and r[110] are the complex reflectance along
the directions [¯110] and [110] denoted x and y respec-
tively. The y direction is defined along the silicon dimer
rows. The experimental spectrum of the clean surface has
been measured on a nominal (100) surface prepared by
the procedure described in Ref.21 and has been scaled
to have the same total anisotropy as the spectrum ob-
tained by Jaloviar et al38
. Our procedure, contrary to
Jaloviar’s one, does not achieve a single domain orienta-
tion; the surface contains also minority domains in the
perpendicular direction, decreasing the total anisotropy.
B. Computational details
Calculations have been carried out within the Car-
Parrinello approach23
in the framework of the Density
Functional Theory (DFT)24
, using gradient corrections25
(tests have been also performed using the local-density
approach). The electronic wavefunctions were expanded
in plane waves with an energy cutoff of 45 Ry. The
p(2×2) Si(001) surface was modelled with periodically
repeated slabs containing eight Si layers and a vacuum
region of at least 6.2 ˚A. A monolayer of hydrogen atoms
was used to saturate the dangling bonds on the lower
surface of the slabs. Ethylene molecules were added
on the silicon slab and the system was fully relaxed to-
wards the minimum energy configuration. Structural re-
laxations of the atomic coordinates were performed using
the method of direct inversion in the iterative subspace26
.
The optimization procedure has been repeated several](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-106-320.jpg)




![ANNEXE6
Fully polarization resolved X-ray absorption spectroscopy
of C2H4 on single-domain Si(0 0 1)-(2 Â 1)
Franz Hennies a
, Alexander F€oohlisch a,*, Wilfried Wurth a
, Nadine Witkowski b
,
Mitsuru Nagasono c
, Maria Novella Piancastelli d
a
Institut f€uur Experimentalphysik, Universit€aat Hamburg, Luruper Chaussee 149, D-22761 Hamburg, Germany
b
Laboratoire dÕOptique des Solides, UMR CNRS 7601, Universitee Pierre et Marie Curie, 4 Place Jussieu, 75252 Paris 05, France
c
Department of Materials Science and Engineering, Kyoto University, Kyoto 606-8501, Japan
d
Department of Chemical Sciences and Technologies and INFM, University Tor Vergata, Rome, Italy
Received 29 October 2002; accepted for publication 15 January 2003
Abstract
We present a polarization resolved near edge X-ray absorption fine structure (NEXAFS) investigation of ethylene
(C2H4) adsorbed on the oriented single-domain Si(0 0 1)-(2 Â 1) surface.
From the detected resonances and their polarization dependences C2H4 is found to be strongly bound to the silicon
dimers with the carbon atoms in a sp3
-hybridized state. The molecular axis is rotated around the surface normal with
respect to the dimer axis.
Ó 2003 Elsevier Science B.V. All rights reserved.
Keywords: X-ray absorption spectroscopy; Chemisorption; Silicon; Alkenes
1. Introduction
The adsorption of molecules on silicon surfaces
has been the focus of many investigations over the
last few years. The bonding of hydrocarbons to the
Si(0 0 1)-surface is of particular interest due to the
proposed technological potential of the hydrocar-
bon–silicon interface in bio-molecular sensors or
even future integrated circuitry beyond todayÕs
silicon oxide based semiconductor manufacturing
[1,2].
The Si(0 0 1)-surface exhibits a reconstruction
characterized by dimer formation [3]. Pairs of
neighboring surface atoms form surface dimers
saturating one of their dangling bonds. The still
high reactivity of this surface originates from the
remaining unsaturated bond per surface atom.
Below 200 K a c(4 Â 2)-surface reconstruction is
observed, formed of alternating flipped buckled
dimers. At temperatures above 200 K these buck-
led dimers oscillate resulting in an average (2Â1)-
pattern [4].
The interaction of C2H4 with Si(0 0 1) was first
examined more than 10 years ago [5], leading to a
bonding model where the C2H4 develops a di-r
bond to the dangling bonds of the Si dimers
(Fig. 1). The C2H4 looses its double-bond and the
*
Corresponding author. Tel.: +49-40-8998-3122; fax: +49-
40-8998-2179.
E-mail address: alexander.foehlisch@desy.de (A. F€oohlisch).
0039-6028/03/$ - see front matter Ó 2003 Elsevier Science B.V. All rights reserved.
doi:10.1016/S0039-6028(03)00079-7
Surface Science 529 (2003) 144–150
www.elsevier.com/locate/susc](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-111-320.jpg)
![carbon atoms rehybridize to an sp3
-state. Upon
adsorption the dimer dangling bonds are saturated
and the buckling of the silicon dimers is removed
[6]. At room temperature adsorption of C2H4 leads
to a saturation coverage of one molecule per sur-
face dimer [7].
Although these results have been supported by
many other studies [8–18] since then, about one
central issue no consent has been achieved until
recently: Do the surface dimers stay intact upon
adsorption of C2H4 or is the dimer cleaved with
new dangling bonds appearing in the adsorption
system? The latter model found strong support
over some time, driven by the wish to explain the
affinity of the C2H4-covered silicon surface to the
coadsorption of atomic hydrogen without major
changes in the initial adsorbate complex [9–11].
However, with the latest theoretical [16] and ex-
perimental results the intuitively convincing di-
merized adsorption structure with the ethylene
saturating the surface dangling bonds seems to
be agreed upon. Experimental support for this
bonding configuration is given among others by
photoelectron diffraction measurements [6] and
valence band photoemission (UPS) [12], where
surface states resulting from the dangling bonds
could no longer be detected. Theoretical investi-
gations [15,16,19–24] quite consistently found the
dimerized structure most stable.
With the UPS examination [12] and corre-
sponding density functional calculations (DFT)
[13] a more detailed adsorption geometry of C2H4
to Si(0 0 1) has been proposed. While the question
of bonding geometries has been dominating the
debate for many years, the last cited investigations
focus on the electronic structure of this adsorbate
system. The authors of the UPS study detected a
1D-band-like dispersion of two valence states. The
DFT calculations support the interpretation of
a delocalized one-dimensional adsorbate band
structure along the dimer rows in C2H4 chemi-
sorbed on Si(0 0 1)-(2 Â 1). Lateral overlap of the
1b3u- and 1b2g-derived orbitals, which carry the
bond to the hydrogen atoms, causes Pauli repul-
sion of the respective wave functions. By an 11.4°
rotation of the C–C-bond axis against the dimer
axis around the surface normal and a further dis-
tortion of the molecule the Pauli repulsion is re-
duced and delocalized bands are formed.
These interesting new results demand for a
further investigation of both the occupied and
unoccupied valence structure in order to under-
stand the complete spatial distribution of the va-
lence states. A previous NEXAFS study [25,26]
showed general trends in the modification of the
unoccupied valence states of C2H4 upon adsorp-
tion to Si(0 0 1). Since this investigation had been
performed on a nominal plain (0 0 1)-surface with
a mixing of (2 Â 1)- and (1 Â 2)-domains, the lat-
eral adsorption directions have not been resolved
therein.
We here present a fully polarization resolved
NEXAFS investigation, allowing us to decide
about the orientation of the unoccupied valence
orbitals with respect to the substrate. We are
making use of the possibility to prepare an ori-
ented single-domain Si(0 0 1)-(2 Â 1) crystal in or-
der to distinguish between all excitation geometries
with respect to the Si single-domain surface coor-
dinate system. On such a sample the full geomet-
rical information about the orientation of the
unoccupied valence structure can be obtained with
NEXAFS [27].
2. Experimental methods
2.1. Setup
The experiments have been performed on
beamline I511 [28] at MAX-Lab in Sweden. The
beamline is equipped with a UHV-system for
electron spectroscopy and X-ray emission studies
on surfaces and surface adsorbates. A preparation
Fig. 1. Adsorption model of ethylene C2H4 on Si(0 0 1)-(2 Â1).
F. Hennies et al. / Surface Science 529 (2003) 144–150 145](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-112-320.jpg)
![ANNEXE6
chamber operated at a base pressure of low 10À10
Torr is connected to an analysis chamber (at mid
10À11
Torr). The analysis chamber can be rotated
around the beam axis, allowing one to choose the
detection angle freely with respect to the orienta-
tion of the electric field vector.
The samples used were cut from p-doped silicon
with an intentional 5° deviation of the surface
normal from the [0 0 1]-direction to the [1 1 0]-
direction. This miscut (0 0 1)-surface exhibits di-
atomic steps with terraces of 8.5 dimers width in a
single (2 Â 1) domain [29]. We mounted two sam-
ples with a 90° rotation around the surface normal
with grazing incidence of the synchrotron light at
an angle of 7° to the surface plane. This setup al-
lows us to measure with the electric field vector of
the incident light oriented normal to the surface
and in the surface plane normal and parallel to the
dimer axis.
2.2. Sample preparation
Samples were outgassed in the UHV by radia-
tive heating at 800 K for 12 h. The clean surface
was prepared by cycles of sputtering with xenon at
room temperature and at 800 K with subsequent
annealing to 1250 K. The surface order was
checked with LEED and surface cleanliness was
monitored with XPS.
The C2H4 was dosed at room temperature with
a capillary doser at a chamber background pres-
sure of P ¼ 1:3 Â 10À7
Torr for 15 min. These
conditions lead to saturation coverage [30].
2.3. Absorption measurement and data analysis
The X-ray absorption spectra have been taken
with the Scienta SES-200 hemispherical electron
analyzer in constant final state mode. The pass
energy was set to 500 eV. In this setting an Auger
yield is measured in an energy range of 50 eV
around the carbon KLL Auger at 252 eV. Elec-
trons were detected in an angle of 55° with respect
to the polarization vector.
The photon flux was measured simultaneously
by a reference gold mesh. The Auger yield spectra
from the adsorbate system and from the clean
silicon were first divided by the flux. All spectra
were normalized to the flat region below threshold,
before the clean sample spectra have been sub-
tracted from the adsorption system spectra.
A total energy calibration of the photon energy
has been performed making use of first- and sec-
ond-order photons of the monochromator exciting
the Si 2p electrons.
To rule out beam damage during measurements
we compared the carbon 1s XPS-spectra before
and after the X-ray absorption measurements for
shape and intensity, but no difference was ob-
served.
3. Results and discussion
3.1. Experimental results
In Fig. 2(a) and (b) we show the spectra re-
corded with the ~EE-vector oriented close to normal
to the sample surface (z-direction along the [0 0 1]
crystallographic axis) for each of our differently
mounted samples (compare sketch). The incidence
direction of the X-ray beam is grazing to the sur-
face. The sample surface stands vertical, that is
perpendicular to the horizontal polarization plane
of the insertion device. Both spectra show the same
two sharp resonances at 285.3 and 286.5 eV. These
energies were calibrated with XPS as mentioned
above, so this values represent the true energy with
an error of 6 Æ100 meV. In addition, weak ap-
pearances of two more features can be detected. In
this excitation geometry the spectra obtained are
independent of the azimuthal sample orientation
(compare Fig. 2(a) and (b)). This proves the
comparability of the data collected from these two
samples. These data are also similar to those
published in a previous absorption study and
measured in this geometry on a Si(0 0 1)-surface
with two domains [25,26]. This assures the com-
parability of experiments performed on the nomi-
nal plane and on the oriented single-domain
terraced Si(0 0 1)-(2 Â 1)-surface and confirms that
adsorption of ethylene on step-edges is negligible
[31].
The spectrum recorded with the electric field
vector oriented in x-direction, i.e. parallel to the Si
dimer axis (along [1110]), is shown in Fig. 3. These
146 F. Hennies et al. / Surface Science 529 (2003) 144–150](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-113-320.jpg)
![measurements have been obtained from the same
sample as the one in Fig. 2(b). The light is incident
grazing to the surface perpendicular to the Si di-
mer axis. The sample surface is now parallel to the
polarization plane of the synchrotron (compare
sketch). Here we clearly observe a resonance at 291
eV. A smaller sharp resonance can be seen at 285.3
eV. A broad feature around 297 eV is also visible.
Fig. 4 shows the spectrum acquired with the ~EE-
vector aligned in y-direction (along [1 1 0]). These
data now are collected on the same sample as the
spectrum in Fig. 2(a). The polarization therefore is
again parallel to the surface, but in this case per-
pendicular to the Si dimer axis. In this geometry
we detect three features. The peak at 291 eV as
Fig. 2. NEXAFS spectra of C2H4 adsorbed on Si(0 0 1)-(2 Â 1)
at room temperature measured on two samples with their
(2 Â 1) domains rotated 90° with respect to each other. The ~EE-
vector of the incident photon beam is aligned along the Z-axis
(compare sketch). Peak assignment see text.
Fig. 3. NEXAFS spectra of C2H4 adsorbed on Si(0 0 1)-(2 Â 1)
at room temperature. The ~EE-vector of the incident photon beam
is aligned along the X-axis (compare sketch). Peak assignment
see text.
Fig. 4. NEXAFS spectra of C2H4 adsorbed on Si(0 0 1)-(2 Â 1)
at room temperature. The E-vector of the incident photon beam
is aligned along the Y -axis (compare sketch). Peak assignment
see text.
F. Hennies et al. / Surface Science 529 (2003) 144–150 147](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-114-320.jpg)
![ANNEXE6
well as the broad resonance around 297 eV can be
found again. A further feature appears at ap-
proximately 288 eV. At the onset of the X-ray
absorption flank a weak shoulder can be seen.
3.2. Discussion
We start the discussion of our data with the
resonance at 291 eV. This feature is dominant with
x-polarization and can clearly be observed with y-
polarization. Only a very small admixture of this
state can be detected with z-polarization. The oc-
currence of this peak in the spectrum recorded
with x-polarization can therefore be explained
by assigning the feature to excitation into a rÃ
-
resonance. The corresponding bond is therefore
parallel to the x-axis. We can interpret this rÃ
-
resonance as the antibonding orbital correspond-
ing to the C–C bond. The direction of maximum
intensity is given by the direction of the bond.
Here in ethylene adsorbed on Si(0 0 1) the peak
appears at 291 eV, that is 10 eV lower than in gas
phase free ethylene [32]. The position is now
comparable to the rÃ
-resonance (at 291 eV) in free
ethane C2H6 [33] which is sp3
-hybridized. The rÃ
-
resonance in simple hydrocarbons shifts to lower
energies with increasing C–C bond length [34]. The
shift in ethylene upon adsorption to Si(0 0 1) can
therefore be taken as an experimental evidence for
the loss of the C–C p-bond and the rehybridization
of the carbon atoms.
We now have to understand why this resonance
can be clearly seen with y-polarized light as well. If
the bond is totally aligned along the x-axis respec-
tively if the C–C bond is parallel to the Si–Si dimer
bond axis this transition would not be allowed.
UPS investigations [12,13] proposed a slight rota-
tion of the ethylene molecule around the surface
normal due to a lateral interaction of the mole-
cules. This deviation of the C–C axis from the di-
mer orientation can explain the occurrence of the
rÃ
-resonance in spectra recorded with y-polariza-
tion. Without any assumptions our NEXAFS
measurements therefore give the direct experi-
mental evidence for the rotation of the C–C bond
axis against the Si dimer axis around the surface
normal in C2H4 adsorbed on Si(0 0 1). The addi-
tional very small admixture of the rÃ
-resonance
seen in the spectra recorded with polarization
vector normal to the surface, is however most likely
due to experimental restrictions, e.g. the angle be-
tween the macroscopic and microscopic surface
plane on the terraced crystals and the incomplete
polarization due to the grazing incidence of the X-
rays. To check for out of plane tilting, we have also
determined NEXAFS with the ~EE-vector Æ45° off
the surface normal. Here no variation is observed,
which is an indication of in plane orientation.
Next we turn to the discussion of the ‘‘p’’-type
resonances at 285.3 and 286.5 eV which are seen
prominently in Fig. 2(a) and (b). From the ab-
sorption measurements on free C2H4 [32] and on
C2H4 in weakly bound systems [35] one would
expect a very strong single pÃ
-resonance for ori-
entation of the polarization vector perpendicular
to the plane of the molecule. Our spectra instead
show two resonances that are much weaker, even
weaker than the rÃ
-resonance seen in Fig. 3. Here
we can prove that C2H4 has lost its p-bond and is
rehybridized when adsorbed to Si(0 0 1). In the
absorption spectra we can see the antibonding
counterparts of the tetrahedrally oriented sp3
-
bonds, i.e. one C–Si bond directed towards the
surface and two C–H bonds which are now seen to
be lifted upwards. The fourth bond is the C–C r-
bond. The C–H bonds are equivalent and we as-
sign one of the split pÃ
to the antibonding C–Si
state and the other to the C–H2, but there is no
reason for which bond should be assigned to which
resonance in the first place. Our fully polarization
resolved measurements however give a certain way
to identify the bonds in the observed resonances.
The first resonance at 285.3 eV is not only
strongly enhanced with z-polarization but can also
clearly be seen with x-polarization. As a slight
shoulder it occurs very weakly in the spectra re-
corded with y-polarized light. That it is clearly
visible with x-polarization but can hardly be seen
with y-polarization gives a strong indication to
identify this resonance to have predominant C–Si
character. If this bond would be aligned with the Si
dimer it would only be visible with z- and x-
polarization. But since the molecule is rotated (see
above) this explains the observation of this state as
a shoulder in the spectrum recorded with y-
polarization.
148 F. Hennies et al. / Surface Science 529 (2003) 144–150](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-115-320.jpg)
![We then have to assign the second resonance at
286.5 eV to a state with major C–H2 admixture. Its
strong contribution to the spectra recorded with z-
polarization gives an experimental evidence that
this bond is not lying in the molecular plane any
more, but that the H atoms are lifted out of the
plane. This again confirms the sp3
-hybridization.
As a C–H2 bond it then should show up with x- as
well as with y-polarization. Its part in the spectra
measured with x-polarization is difficult to deter-
mine because it lies in the strong rise of the rÃ
-
resonance. Anyhow the flank of the rÃ
-resonance
seems to include the part of another resonance.
With y-polarization this state can clearly be seen,
although it seems to be shifted to around 288 eV.
Here probably the dispersion of the occupied
states related to the C–H2 bond as measured with
UPS [12,13] can be taken into account to explain
this effect.
Finally the broad feature at approx. 297 eV can
be more or less seen in all geometries. We attribute
this state to the ‘‘continuum resonance’’ [36] called
state exhibited by all simple hydrocarbons in the
gas phase, which is caused by multiple excitations.
So far we have discussed our NEXAFS results
assuming that the features result mainly from the
ethylene species adsorbed on the terraces. In
principle there could also be a sizeable contribu-
tion from step species. However, additional mea-
surements at polarization angles of Æ45° with
respect to the surface normal (not shown) which
exhibit no difference indicate that this contribution
must be small.
4. Conclusion
Using vicinal single-domain Si(0 0 1) crystals we
have been able to perform a fully polarization re-
solved NEXAFS study of C2H4 adsorbed on ori-
ented Si(0 0 1)-(2 Â 1).
By investigating the unoccupied states our
measurements not only prove the adsorption of
C2H4 on top and along the Si surface dimers, but
also give direct evidence of a slight rotation of the
C2H4 C–C bond axis around the surface normal
against the dimer axis. In addition we find the C–
H2 bond to be flipped out of the molecular plane
by identifying the z-component of its correspond-
ing antibonding state.
The full picture of the spatial orientation of the
unoccupied states and the shift of the rÃ
-resonance
to lower energy support the bonding model of
C2H4 adsorbed on Si(0 0 1) with sp3
-rehybridiza-
tion of the carbon atoms and formation of a strong
covalent bond to the Si surface dimer atoms.
Acknowledgements
We thank B. Naydenov, A. Fink and W. Widdra
for providing the samples and preparation infra-
structure. We acknowledge support from MAX-
Lab staff during the beamtime. Financial support
was given through the EU access to research in-
frastructure program (ARI), the DFG (grant Fo
343/1-1) and the ‘‘Grant-in-Aid for Japan Society
for the promotion of Science Fellows’’.
References
[1] J.T. Yates, Science 279:5349 (1998) 335.
[2] M. Schulz, Nature 399 (1999) 729.
[3] R. Hamers, R. Tromp, J. Demuth, Phys. Rev. B 34 (1986)
5343.
[4] R.A. Wolkow, Phys. Rev. Lett. 68 (1992) 2636.
[5] J. Yoshinobu, H. Tsuda, M. Onchi, M. Nishijima, J.
Chem. Phys. 87 (1987) 7332.
[6] P. Baumg€aartel, R. Lindsay, O. Schaff, T. Gießel, R.
Terborg, J.T. Hoeft, M. Polcik, A.M. Bradshaw, M.
Carbone, M.N. Piancastelli, R. Zanoni, R.L. Toomes,
New J. Phys. 1 (1999) 20.
[7] C.C. Cheng, R.M. Wallace, P.A. Taylor, W.J. Choyke, J.T.
Yates, J. Appl. Phys. 67 (1990) 3693.
[8] L. Clemen, R.M. Wallace, P.A. Taylor, M.J. Dressler, W.J.
Choyke, W.H. Weinberg, J.T. Yates, Surf. Sci. 268 (1992)
205.
[9] C. Huang, W. Widdra, W.H. Weinberg, Surf. Sci. 315
(1994) L953.
[10] W. Widdra, C. Huang, W. Weinberg, Surf. Sci. 329 (1995)
295.
[11] W. Widdra, C. Huang, S.I. Yi, W. Weinberg, J. Chem.
Phys. 105 (1996) 5605.
[12] W. Widdra, A. Fink, S. Gokhale, P. Trischberger, U.
Gutdeutsch, U. Birkenheuer, N. R€oosch, D. Menzel, Phys.
Rev. Lett. 80 (1998) 4269.
[13] U. Birkenheuer, U. Gutdeutsch, N. R€oosch, A. Fink, S.
Gokhale, P. Trischberger, D. Menzel, W. Widdra, J.
Chem. Phys. 108 (1998) 9868.
[14] A.J. Fisher, P.E. Bl€oochl, G.A.D. Briggs, Surf. Sci. 374
(1997) 298.
F. Hennies et al. / Surface Science 529 (2003) 144–150 149](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-116-320.jpg)
![ANNEXE6
[15] N. Pangher, L. Wilde, M. Polcik, J. Haase, Surf. Sci. 372
(1997) 211.
[16] J.-H. Cho, L. Kleinman, C.T. Chan, K.S. Kim, Phys. Rev.
B 63 (2001) 073306, erratum: Phys. Rev. B 64 (2001)
199902.
[17] M.P. Casaletto, R. Zanoni, M. Carbone, M.N. Piancas-
telli, L. Aballe, K. Weiss, K. Horn, Phys. Rev. B 62 (2000)
17128.
[18] S.H. Xu, M. Keeffe, Y. Yang, C. Chen, M. Yu, G.J.
Lapeyre, E. Rotenberg, J. Denlinger, J.T. Yates, Phys.
Rev. Lett. 84 (2000) 939.
[19] B.I. Craig, P.V. Smith, Surf. Sci. 276 (1992) 174, erratum:
Surf. Sci. 285 (1993) 295.
[20] P.-L. Cao, R.-H. Zhou, J. Phys.: Condens. Matter 5 (1993)
2887.
[21] B.I. Craig, Surf. Sci. 329 (1995) 293.
[22] K.-A. Feng, Z.H. Liu, Z. Lin, Surf. Sci. 329 (1995) 77.
[23] F. Rochet, F. Jolly, F. Boumel, G. Dufour, F. Sirotti, J.L.
Cantin, Phys. Rev. B 58 (1998) 11029.
[24] G.A.D. Briggs, A.J. Fisher, Surf. Sci. Rep. 33 (1999) 1.
[25] F. Matsui, H.W. Yeom, A. Imanishi, K. Isawa, I.
Matsuda, T. Ohta, Surf. Sci. 401 (1998) L413.
[26] F. Matsui, H.W. Yeom, I. Matsuda, T. Ohta, Phys. Rev. B
62 (2000) 5036.
[27] J. St€oohr, NEXAFS Spectroscopy, Springer, Berlin, 1992.
[28] R. Denecke, P. V€aaterlein, M. B€aassler, N. Wassdahl, A.
Nilsson, J.-E. Rubensson, J. Nordgren, N. Maartensson, R.
Nyholm, J. Electron Spectrosc. Relat. Phenom. 101–103
(1999) 971.
[29] S. van Dijken, H.J.W.Z. und B. Poelsema, Surf. Rev. Lett.
5 (1998) 15.
[30] A. Fink, Organische molek€uule auf halbleitern: Adsorption
und elektronische struktur unges€aattigter kohlenwasserst-
offe auf Si(1 0 0), Ge/Si(1 0 0) und Ge(1 0 0) oberfl€aachen,
Ph.D. Thesis, TU M€uunchen, 2001.
[31] A. Fink, W. Widdra, W. Wurth, C. Keller, M. Stichler, A.
Achleitner, G. Comelli, S. Lizzit, A. Baraldi, D. Menzel,
Phys. Rev. B 64 (2001) 045308.
[32] R. McLaren, S.A.C. Clark, I. Ishi, A.P. Hitchcock, Phys.
Rev. A 36 (1987) 1683.
[33] A.P. Hitchcock, I. Ishii, J. Electron Spectrosc. Relat.
Phenom. 42 (1987) 11.
[34] J. St€oohr, F. Sette, A. Johnson, Phys. Rev. Lett. 53 (1984)
1684.
[35] H. Rabus, D. Arvanitis, M. Dohmke, K. Baberschke, J.
Chem. Phys. 96 (1992) 1560.
[36] S. Sorensen, M. Wiklund, S. Sundin, A. Ausmees, A.
Kikas, S. Svensson, Phys. Rev. A 58 (1998) 1879.
150 F. Hennies et al. / Surface Science 529 (2003) 144–150](https://image.slidesharecdn.com/93e9fa75-1ab7-448c-b5af-d4ade6a17012-160518114906/85/habilitation-117-320.jpg)