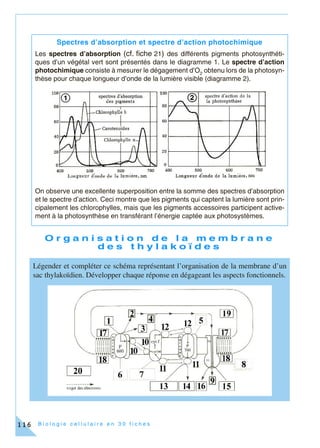
Cet ouvrage, destiné aux étudiants en biologie cellulaire, propose une approche moderne incluant des méthodes expérimentales en 30 fiches thématiques. Chaque fiche couvre des concepts clés et des techniques associés, intégrant des exercices pratiques basés sur de véritables expériences de laboratoire. L'objectif est de former des étudiants à une compréhension approfondie et pratique de la biologie cellulaire, en reflétant l'évolution des connaissances et des outils pédagogiques.



























































































































































































![Biologie cellulaire1[1].pptx...............](https://cdn.slidesharecdn.com/ss_thumbnails/biologiecellulaire11-251218173415-db5e4bf0-thumbnail.jpg?width=640&height=640&fit=bounds)




















































