Téléchargé 58 fois












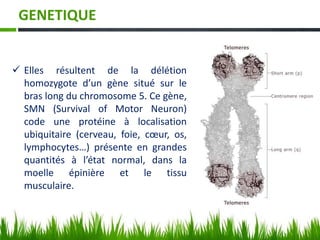


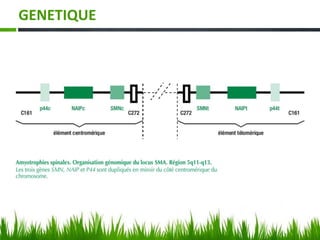







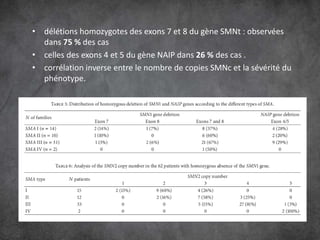











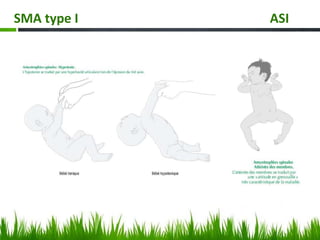
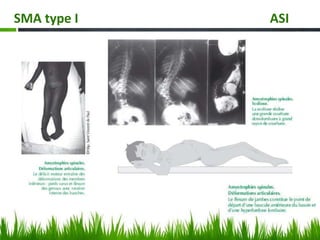















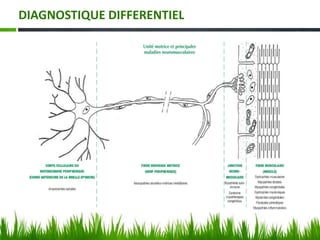




















Les amyotrophies spinales (SMA) sont un groupe de maladies neuromusculaires héréditaires caractérisées par la dégénérescence des motoneurones, avec des présentations cliniques variées. La maladie est principalement causée par des anomalies génétiques, notamment des délétions du gène SMN sur le chromosome 5, et a des implications importantes sur la fonction respiratoire et orthopédique. Le diagnostic repose sur l'évaluation clinique et peut être confirmé par des tests génétiques.















































