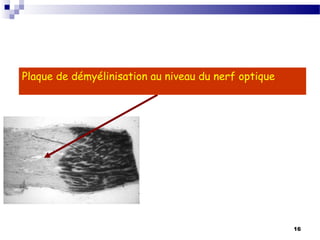
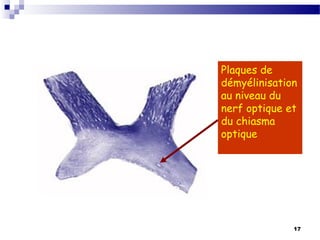
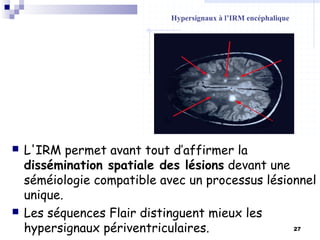
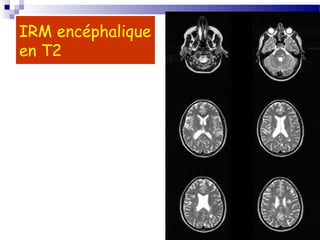
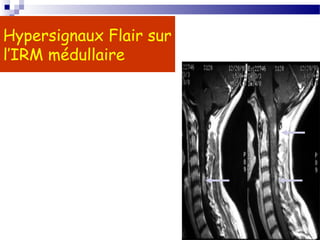
La sclérose en plaques (SEP) est une maladie neurologique fréquente, souvent débutant chez l'adulte jeune, caractérisée par des poussées cliniques de démyélinisation dans le système nerveux central. Sa prévalence varie selon les régions, avec environ 40 000 cas en France, et plusieurs facteurs, notamment génétiques et environnementaux, contribuent à son étiologie encore mal comprise. Le diagnostic repose sur la dissémination des lésions et des examens paracliniques tels que l'IRM et l'analyse du liquide céphalorachidien.
































































![Comas 3eme année [lecture seule]](https://cdn.slidesharecdn.com/ss_thumbnails/comas3emeannelectureseule-130912063627-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




































![Cours d immunologie generale Dr Louis [Enregistrement automatique].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/coursdimmunologiegeneraledrlouisenregistrementautomatique-260114234321-8e6b96ef-thumbnail.jpg?width=640&height=640&fit=bounds)




