Téléchargé 142 fois



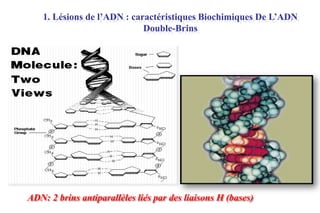

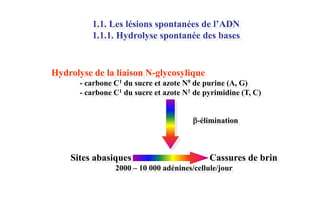
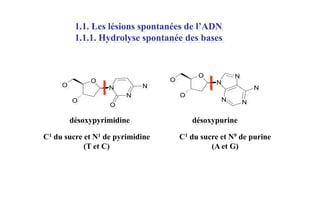
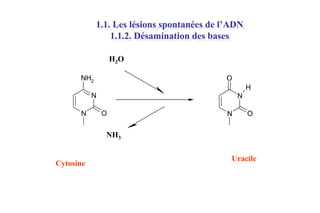




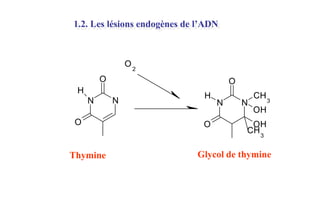


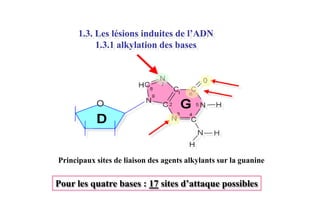

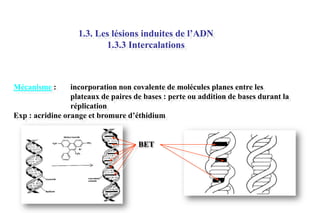

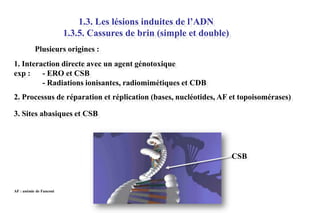











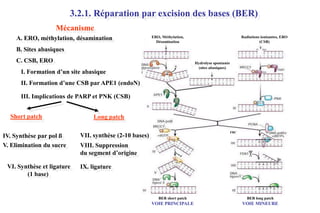


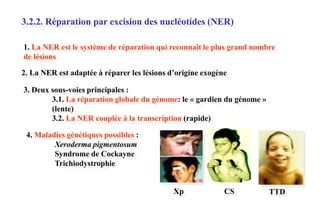
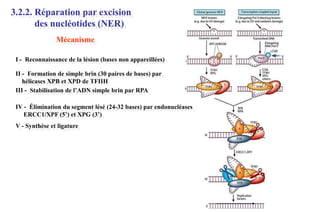

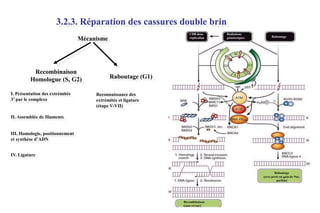


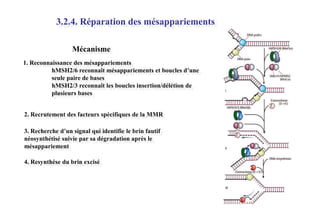


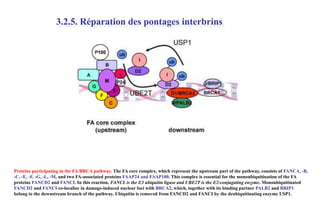
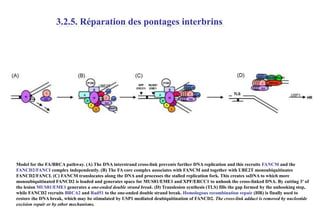

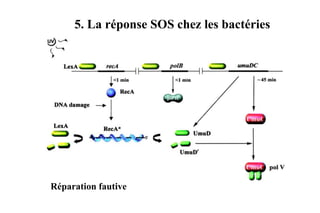
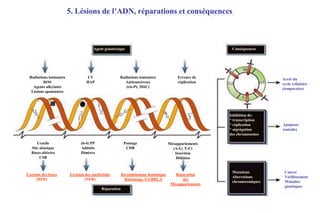

Le document traite des lésions et mutations de l'ADN, ainsi que des mécanismes de réparation associés. Il décrit les différentes causes de lésions, qu'elles soient spontanées, endogènes ou induites par des agents environnementaux, et les conséquences potentiellement graves de ces dommages, allant des mutations génétiques aux cancers. Enfin, il présente les divers systèmes de réparation de l'ADN qui peuvent corriger ces altérations, soulignant l'importance de ces mécanismes pour maintenir l'intégrité de l'information génétique.















































