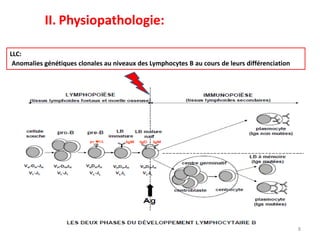
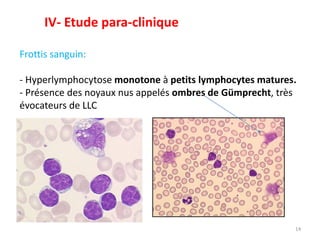
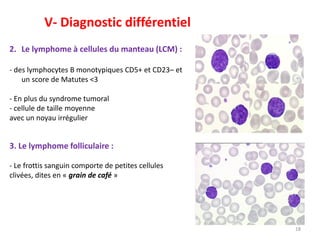
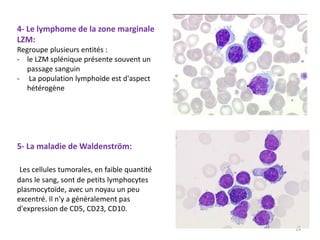
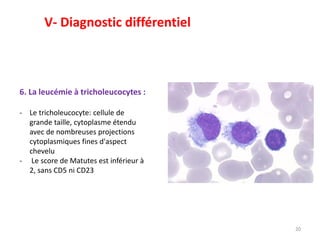
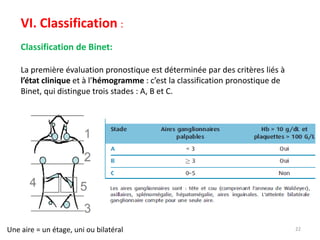
La leucémie lymphoïde chronique (LLC) est une prolifération lymphoïde B monoclonale fréquente chez les adultes, souvent asymptomatique au moment du diagnostic. Le diagnostic repose sur l'hémogramme, le frottis sanguin et l'immunophénotypage, tandis que le traitement varie selon le stade. Les complications incluent des infections et des troubles auto-immuns, rendant une prise en charge précoce essentielle.