
Recommandé
Contenu connexe
Similaire à s-m-c-5-m30-chimie-th-orique-el-issami.pdf
Similaire à s-m-c-5-m30-chimie-th-orique-el-issami.pdf (20)
Dernier
Dernier (7)
s-m-c-5-m30-chimie-th-orique-el-issami.pdf
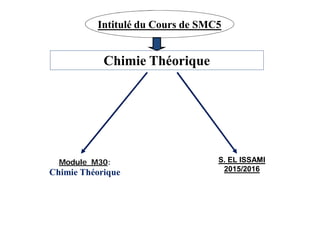
- 1. Chimie Théorique Module M30: Chimie Théorique S. EL ISSAMI 2015/2016 Intitulé du Cours de SMC5
- 2. 1 1ère ère Partie Partie Rappel Chapitre I : Le Moment cinétique Chapitre II : L’atome d’hydrogène et Systèmes hydrogénoïdes Chapitre III : Les méthodes d’approximations Chapitre IV : Atome à plusieurs électrons Chapitre V : Les Termes Spectraux SOMMAIRE
- 3. Rappel Limites de La Mécanique Classique :Etude des objets infiniment petits: Expériences I. a. Le rayonnement du corps noir b. L’effet photoélectrique c. L’effet Compton d. La diffraction des électrons II. Le spectre des atomes et molécules a. Le modèle de Bohr de l’atome d’hydrogène b. L'hypothèse de L. de Broglie et la dualité onde/corpuscule
- 4. •Les phénomènes quantiques ont une nature aléatoire. On ne peut prévoir les résultats d’une expérience que sous forme statistique (grand nombre d’événements) ou probabiliste (un seul évènement). • Certaines grandeurs physiques, qui classiquement peuvent prendre un ensemble continu de valeurs n’adoptent en mécanique quantique que des valeurs discrètes (ex : niveaux d’énergie des atomes, projection du moment magnétique de l’électron sur un axe). Naissance de la Mécanique Quantique
- 5. •Nous devons abandonner la notion de trajectoire et de la substituer par la notion de fonction d’onde •(r, t) et de probabilité de présence. • Le mouvement d’une particule microscopique ne peut se décrire en terme de trajectoire mais en terme de probabilité. •Toutes les informations sur une particule microscopique à l’instant t donné sont contenu dans une onde décrite par une fonction (r, t) appelée fonction d’onde. La probabilité pour que la particule se manifeste à la position r à l’ instant t donné est exprimé par : • |Ψ(r, t)|2
- 6. Max Born a postulé en 1924 que la probabilité pour que la particule soit trouvée a l’instant t dans un élément de volume infinitésimal dv = dx dy dz centré en un point M de coordonnées x, y, z est : dp( r, t) = |Ψ(r, t)|2 dv où |Ψ(r, t)|2= Ψ*(r, t) Ψ(r, t) est la densité de probabilité. Si le système est constitué d’une seule particule, la probabilité de trouver celle-ci en un point quelconque de l’espace à l’instant t est une certitude, donc par convention , égale à l’unité: P= |Ψ(r, t)|2 dv = 1 Pour représenter un état physique, la fonction d’onde doit donc être « de carrée sommable »; càd une fonction dont l’intégrale converge |Ψ(r, t)|2 dv.
- 7. 1ère Partie
- 8. 1 1ème ème Partie Partie 8 Chapitre I Le moment Cinétique I. Introduction L’opérateur moment cinétique joue un rôle très important en mécanique quantique, non seulement dans le cas des systèmes soumis à un potentiel central pour lesquels il permet de classer les états, mais aussi parce qu’il existe un moment cinétique intrinsèque des particules, le spin, qui n’a pas d’équivalent classique. I.1/ Le moment cinétique orbital ( ) L̂ Le moment cinétique orbital ou moment angulaire est un opérateur qui possède un équivalent en mécanique classique, il est défini par la relation : L̂ = r̂ p̂ Vecteur position Vecteur quantité de mouvement
- 9. 9 Ce moment cinétique orbital est dû à la rotation de l’électron autour du noyau. Celui-ci n’a pas d’équivalent en mécanique classique. Il est dû à la rotation intrinsèque de l’électron autour de lui-même. C’est une grandeur purement quantique. I.2/ Le moment cinétique de spin ( ) Ŝ I.3/ Le moment cinétique total ( ) Jˆ Pour un système constitué de particules en rotation, le moment cinétique total (ou moment angulaire total) « » peut être défini comme suit : J ˆ J = L + S = i i l + i i s ; L = i i l J = i i j ; i i i s l j ; S = i i s
- 10. 10 II. / Moment cinétique orbital II.1./ Définition a- En mécanique classique, le moment cinétique (L ) d’une particule d’impulsion p placé en un point M situé à la distance r de l’origine o est : L = r Λ p ; p = mV r = Vecteur position O M r p L x y z r = m V m P z y x ; z y x
- 11. 11 L Pz Py Px z y x Lz Ly Lx = L où d yPx xPy xPz zPx zPy yPz ' yPx xPy Lz xPz zPx Ly zPy yPz Lx = b- En mécanique quantique : il suffit de remplacer r et p par leur forme opérationnelle (opérateur correspondant) pour obtenir l’expression de l’opérateur moment cinétique orbital. rˆ z y x ˆ ˆ ˆ = i i P ˆ ˆ z y x ˆ ˆ ˆ ;
- 12. 12 L̂ p r ˆ ˆ L̂ z y x i z y x x y y x z x x z y z z y i = et devient : En coordonnées cartésiennes les composantes de l’opérateur sont donc : ) ( ˆ ) ( ˆ ) ( ˆ x y y x i z L z x x z i y L y z z y i x L L̂
- 13. 13 On notera l’opérateur scalaire : L̂ = L̂ . L̂ = 2 2 2 ˆ ˆ ˆ z y x L L L 2 L̂ 2 II.2./ Relation de commutation 1/- Composantes du moment cinétique : x L̂ y L̂ z L̂ Les trois composantes du moment cinétique orbital: ne commutent pas. On trouve par un calcul simple : [ x L̂ , y L̂ ] = z L i ˆ ; [ y L̂ , z L̂ ] = x L i ˆ et [ z L̂ , x L̂ ] = y L i ˆ Dans le cas de la première relation on a : [ x L̂ , y L̂ ] = x L̂ . y L̂ - y L̂ . x L̂
- 14. 14 x L̂ . y L̂ = - ћ2 [y. ] . . . . . . . z x y z x z y z z x z y x z z y L̂ . x L̂ = - ћ2 [z. y z z x z y z x y z x z z y x . . . . . . . . . ] D’où [ x L̂ , y L̂ ] = - ћ2 [y. y x x ] z L i ˆ 1 [ x L̂ , y L̂ ] = z L i ˆ 0 Remarques : les opérateurs x L̂ , y L̂ et z L̂ ne commutent pas Ces opérateurs n’ont pas le même système de fonctions propres. Du point de vue physique, il est impossible de mesurer simultanément les 3 composantes du moment cinétique.
- 15. 15 2/- L’opérateur : cet opérateur commute avec les composantes de en effet : 2 L̂ L̂ [ 2 L̂ , x L̂ ] = 2 L̂ . x L̂ - x L̂ . 2 L̂ 2 L̂ = 2 2 2 ˆ ˆ ˆ z y x L L L [ 2 L̂ , x L̂ ] = [ x L L L L z y x ˆ , ˆ ˆ ˆ 2 2 2 ] = [ ] ˆ , ˆ [ ] ˆ , ˆ [ ] ˆ , ˆ 2 2 2 x L L x L L x L L z y x = [ ] ˆ , ˆ . ˆ [ ] ˆ , ˆ . ˆ [ ] ˆ , ˆ . ˆ x z z x y y x x x L L L L L L L L L
- 16. 16 Or [ C B A ˆ , ˆ . ˆ ] = A [ C B ˆ . ˆ ] + [ C A ˆ . ˆ ] B̂ [ 2 L̂ , x L̂ ] = x L̂ [ x L x L ˆ , ˆ ] + [ x L x L ˆ , ˆ ] y L x L ˆ ˆ [ x L y L ˆ , ˆ ] + [ x L y L ˆ , ˆ ] y L̂ + z L̂ [ x L z L ˆ , ˆ ] + [ x L z L ˆ , ˆ ] z L̂ = y L̂ (- ih z L̂ ) + (-ih z L̂ ) y L̂ + z L̂ (ih y L̂ ) + (+ih y L̂ ) z L̂ . = (- ih) [ y L̂ . z L̂ + z L̂ . y L̂ - z L̂ . y L̂ - y L̂ . z L̂ ] = 0 [ 2 L̂ , x L̂ ] = 0 De même on peut vérifier que : [ 2 L̂ , y L̂ ] = 0 et [ 2 L̂ , z L̂ ] = 0 0 ] ˆ , ˆ [ 0 ] ˆ , ˆ [ 0 ] ˆ , ˆ [ 2 2 2 z L L y L L x L L
- 17. 17 de point de vue physique il est possible de mesurer simultanément 2 L̂ et une composante de L̂. D’après ces relations de commutation, il est possible de construire un système de fonction propre commun à 2 L̂ et à l’une des composantes de L̂ . On peut donc former un E.C.O.C (ensemble complet d’observables qui commutent). Particulièrement adapté aux calculs faisant intervenir le moment cinétique constitué du carré ( 2 L̂ ) et de l’une des composantes de L̂ .
- 18. 18 Par convention, on choisit l’E.C.O.C z L L ˆ , ˆ2 . Les états propres communs à ces deux opérateurs sont notés : . , , m l ou m l l, m sont des nombres quantiques définies de manière à ce que les valeurs propres de 2 L̂ et z L̂ , soient respectivement : m z L l l L ˆ ) 1 ( ˆ 2 2 Autrement dit, on pose : Les équations aux valeurs propres de z 2 L̂ et L̂ m l m m l z L , , ˆ avec l ≥ 0 et -l ≤ m ≤ l 2 L̂ m l l l m l , ) 1 ( , 2
- 19. 19 III/. Généralisation On réserve le symbole L̂ au moment cinétique orbital, Ŝ au moment cinétique de spin. On appellera opérateur moment cinétique, noté Ĵ tout opérateur hermétique dont les composantes satisfont les relations de commutation : [ z J i y J x J ˆ ] ˆ , ˆ ; x J i z J y J ˆ ] ˆ , ˆ [ y J i x J z J ˆ ] ˆ , ˆ [ et 0 ] ˆ , ˆ [ 2 J J y J x J ˆ , ˆ ou z Ĵ
- 20. 20 IV. Les opérateurs escalateurs Soit L̂ et L̂ des opérateurs non hermétiques définis sous la forme de combinaison linéaire des opérateurs x L̂ et y L̂ . Les opérateurs L et L ˆ ˆ sont adjoint l’un de l’autre. ) ˆ ( ) ˆ ( L L i L L y L L i L L L L x L y L i x L L y x 2 ˆ ˆ ˆ ˆ ˆ ˆ 2 ˆ ˆ ˆ ˆ ˆ ˆ
- 21. 21 1./. Relation de commutation entre L L ˆ , ˆ et les composantes de L̂ . ] ˆ ˆ , ˆ [ ] ˆ , ˆ [ y x z z L i L L L L = ] ˆ , ˆ [ ] ˆ , ˆ [ y z x z L L i L L = x y L L i ˆ ˆ = ) ˆ ˆ ( y x L i L = L̂ L̂ 0 ˆ ] ˆ , ˆ [ L L z L de même[ ] ˆ , ˆ L Lx = - ћ 0 ˆ z L et [ ] ˆ , ˆ L Ly = -i ћ 0 ˆ z L Pour L̂ on trouve également : 0 ˆ ] ˆ ˆ , ˆ [ ] ˆ , ˆ [ z y x x x L L i L L L L z y L i L L ˆ ] ˆ , ˆ [ L i L Lz ˆ ] ˆ , ˆ [
- 22. 22 2./. Relation de commutation entre , et 2 L̂ L̂ L̂ * ] ˆ ˆ , ˆ [ ] ˆ , ˆ [ 2 2 y L i L L L L x ; 2 2 2 2 ˆ ˆ ˆ ˆ z y x L L L L = 0 ] ˆ , ˆ [ ] ˆ , ˆ [ 2 2 y x L L i L L 2 L̂ commute avec les composantes de L̂, en effet : 0 ] ˆ , ˆ [ 0 ] ˆ , ˆ [ ; 0 ] ˆ , ˆ [ 2 2 2 z L L et L L L L y x 0 ] ˆ , ˆ [ 2 L L * 0 ] ˆ , ˆ [ ] ˆ , ˆ [ ] ˆ , ˆ [ ] ˆ ˆ , ˆ [ ] ˆ , ˆ [ 2 2 2 2 2 L L L L i L L L i L L L L y x y x
- 23. 23 3./. Relation de commutation entre , L̂ L̂ L L L L L L ˆ . ˆ ˆ . ˆ ] ˆ , ˆ [ * x y y x y x y x y x L L i L L i L L L i L L i L L L ˆ ˆ ˆ ˆ ˆ ˆ ) ˆ ˆ )( ˆ ˆ ( ˆ ˆ 2 2 = 2 2 ˆ ˆ y x L L i [ x L̂ , y L̂ ] = 2 2 ˆ ˆ y x L L + z L̂ Or 2 2 2 2 ˆ z y x L L L L L̂ L̂ z z L L L ˆ ˆ ˆ 2 2
- 24. 24 * L̂ L̂ ) ˆ ˆ )( ˆ ( y x y x L i L iL L = 2 2 ˆ ˆ y x L L +i [ x L̂ , y L̂ ] = 2 2 ˆ ˆ y x L L z L̂ L̂ L̂ z y x L L L ˆ ˆ 2 2 , L̂ L̂ z z L L L ˆ ˆ 2 2 d’où [ L L ˆ , ˆ ] = z L̂ 2 0 les opérateurs L̂ et L̂ ne commutent pas .
- 25. 25 V. Valeurs et vecteurs propres des opérateurs constituants un E.C.O.C 1./ Cas de l’E.C.O.C et 2 L̂ z L̂ E.C.O.C : ensemble complet d’observable qui commute. [ 2 L̂ , z L̂ ] = 0 2 L̂ et z L̂ commutent auront le même système de fonction propres. Soit une fonction propre commune à 2 L̂ et z L̂ . m L k L z ˆ ˆ 2 2 2 2 2 k et m désigne respectivement les valeurs propres associée à 2 L̂ et z L̂ . Toute démonstration permet d’établir la relation entre les deux valeurs propres.
- 26. 2 h ; avec h: constante de Planck : 6,626 10-34 J.S (u.a) 1 Dans le système d’unité atomique, la valeur propre associé à l’opérateur est : k2 et celle de l’opérateur est : m. La relation entre les deux est : l(l+1) = k2 2 L̂ z L̂ Il existe (2l + 1) valeurs de m. l = 2 ; -2 m +2 m = -2 , -1 , 0 , 1 , 2 2 x 2 + 1 = 5 val. de m. 5fonctions d’état l,m l 0 et -l m +l Ex. : l = 1 -1 m 1 m = 0 , 1 2 x 1 + 1 = 3 valeurs de m. 3 fonctions d’état l,m
- 27. 27 Les valeurs propres des opérateurs z L et L ˆ ˆ2 2 L̂ z L̂ ћ2 l(l+1) ћm 1-c/. Généralisation Ŝ Ĵ Soit un opérateur moment cinétique quelconque : = ou = ou = + Ĵ Ĵ L̂ L̂ S ˆ Ĵ m j, désigne la base propre commune au couple J2 et JZ , on a alors la relation suivante : m j j j h m j j , ) 1 ( , ˆ 2 2 m j hm m j jZ , , ˆ
- 28. 28 2./ Valeurs propres associées à et L̂ L̂ Donc 1 , , 1 , , ) 1 ( ) 1 ( ˆ ) 1 ( ) 1 ( ˆ m l m l m l m l m m l l L m m l l L Si m= l 0 ) 1 ( ) 1 ( ˆ 1 , , l l l l l l l l L Si m = -l 0 ) 1 ( ) 1 ( ˆ 1 , , l l l l l l l l L
- 29. 29 VI. Les fonctions propres des opérateurs z L et L ˆ ˆ2 1./ Le moment cinétique dans les systèmes de coordonnées 1.a./ En coordonnées cartésiennes ) ( ˆ ) ( ˆ ) ( ˆ x y y x i L z x x z i L y z z y i L z y x 2.b./ En coordonnées sphériques x = r sinθ cos y = r sinθ sin z = r cosθ r ≥ 0 ; 0 θ π ; 0 2π
- 31. 31 2./. Fonctions propres de z L̂ i Lz ˆ On cherche les solutions l,m fonctions propres des deux variables θ En effet, z L̂ dépend d’une seule variable () mais 2 L̂ dépend de 2 variables (θ et ) et l,m commune à 2 L̂ et z L̂ . z L̂ l,m (θ,) = ћm l,m(θ,) ) , ( ) , ( , , m l m l m i l,m(θ,) = θ(θ).() ) ( ) ( ) ( ) ( m i ) ( ) ( ) ( ) ( m i Equation aux valeurs propres de z L̂ LZ
- 32. 32 ) ( ) ( m i imd ) ( ) ( () im Ne ; N : est une constante de Normalisation 1 ) ( ) ( 2 2 1 o im im d e e N ; N = 2 1 1 im e 2 1 ) ( ; m est un entier
- 33. 33 3./. Fonctions propres de 2 L̂ ) , ( ) 1 ( ) , ( ˆ , 2 , 2 m l m l l l L ) ( ) ( ) 1 ( ) ( ) ( ˆ 2 2 l l L -ћ2 ) ( ) ( 2 sin 1 1 2 2 2 2 tg = ћ2 l(l+1) θ() () )! ( )! ( 2 ) 1 2 ( ) ( , m l m l l m l 1/2 Pl m (cos)
- 34. 34 Polynôme de Légendre est : Pl(cos) = l l l l d d l ) 1 (cos ) cos ( ! 2 1 2 Les polynômes de Légendre associés sont liés aux polynômes de Légendre par : Pl m (cos) = (1 – cos2 )m/2 ) (cos ) cos ( l m P d m d La solution générale de l’équation globale en θet est noté : l,m(,) ou ) , ( m l est appelée harmonique sphérique. l m l m l l m l d d m l m l l l 2 , ) (sin cos ) (sin )! ( 4 )! )( 1 2 ( ! 2 ) 1 ( ) , ( eim
- 35. 35 Application : l=0 , m=0 4 1 ) , ( , o o l=1 ,m = 1 0 1 ) (sin cos 4 3 2 1 2 0 , 1 d d cos 4 3 ) , ( 0 , 1
- 36. 36 2 2ème ème Partie Partie 36 Chapitre II Atome d’hydrogène et systèmes hydrogénoïdes Hydrogénoïde : c’est un ion ayant une charge nucléaire égale à +Ze et qui possède un seul électron qui gravite autour du noyau . On va étudier l’atome d’hydrogène car c’est le plus simple des systèmes hydrogénoïdes. On résoudra l’équation de shrödinger de manière analytique exacte. I./. Résolution de l’équation de Schrödinger I.1./- Hamiltonien du système
- 37. 37 On cherche à décrire le mouvement interne de ce système constitué de deux particules, c'est à dire leur mouvement par rapport au centre de gravité de l'atome. En raison de la très grande différence de masse entre les deux particules, on peut considérer que le centre de gravité de l'ensemble est confondu avec le noyau. On ne considère alors que le mouvement de l'électron par rapport au noyau supposé fixe et pris comme origine d'un référentiel "atomique". Les systèmes hydrogénoïdes sont les seuls systèmes pour lesquels il est possible de déterminer les solutions exactes de l'équation de Schrödinger. Pour les systèmes plus complexes (plus de deux particules en interaction), on ne peut déterminer que des fonctions d'onde approchées.
- 38. 38 N e M V m H 2 ˆ 2 ˆ 2 2 ; V : est le potentiel associé à l’énergie d’attraction éléctron -noyau Or : M >> me M 1 << m 1 TN << Te : Hamiltonien du système constitué de l’e- et du noyau. D’où H ˆ m 2 2 e + V̂ = el Ĥ On a fait une approximation. C’est l’approximation de BORN- OPPENHEIMER c'est-à-dire l’énergie cinétique du noyau est négligeable devant celle de l’e- . Elle appelée approximation des noyaux fixes ou approximation adiabatique. Énergie cinétique du noyau est négligeable devant celle de l’e- Te TN
- 39. 39 r Ze r qq dr r qq Fdr V 0 2 0 2 0 4 4 ' 4 ' 1 4 0 Dans un système d’unité donnée Pour simplifier les expressions mathématiques et faire disparaître les constantes, on introduit les unités atomiques (u. a.) de longueur et d'énergie. 1 u.a = a0= 0,529Å 1 u.a = 27,21 eV Compte tenu de ce nouveau système d'unités, on peut réécrire l'opérateur hamiltonien de manière plus simple : r z H 2 1 ˆ 1 me =1 et e =1
- 40. 40 r Ze V 2 ˆ 2 2 2 2 2 2 2 2 2 2 2 ) ( 2 ˆ z y x Ze z y x m H En coordonnés sphérique on a : 2 2 2 2 2 2 2 sin 1 ) (sin sin 1 ) ( 1 r r r r r r r Ze r r r r r r m H 2 2 2 2 2 2 2 2 2 sin 1 ) (sin sin 1 ) ( 1 2 ˆ L2 D
- 41. 41 r e L D mr H 2 2 2 ) ( 2 1 ˆ 2 2 ) ( r r r D 2 2 2 2 2 sin 1 ) (sin sin 1 L atome d’hydrogène z=1 2 L̂ et Ĥ On peut montrer que commutent
- 42. 42 HL2 = 2 2 2 2 2 ) ( 2 1 L r e L L D mr 0 , 2 L H L2 H = 2 2 2 2 2 ) ( 2 1 L r e L D L mr H, L2 , Lz commutent donc Ils vont avoir le même système de fonctions propres. I.2./- Résolution de l’équation de Schrödinger électronique Equation à résoudre est : H = E On peut écrire : = R(r) () () = R(r) (,) R(r) : fonction radiale (,) : fonction angulaire
- 43. 43 L2 (,) = l(l+1) (,) L’équation est : m 2 2 E r e r r r r r r 2 2 2 2 2 2 2 2 ) sin 1 ) (sin sin 1 ) ( 1 ( = R(r) () ) = R(r) (, ) - L2 ) , ( * ) ( ) ( 2 - 2 2 2 r R r r r mr E r e mr 2 2 2 2 2 2 ) , ( ) sin 1 ) (sin sin 1 R(r) * 2 X ) , ( ) ( 2 - 2 2 r R mr
- 44. 44 ) 1 ( ) , ( sin 1 ) (sin sin 1 ) , ( 1 ) ( 2 ) ) ( ( ) ( 1 2 2 2 2 2 2 2 2 l l E r e mr r r R r r r R ) ( ) 1 ( ) ( ) ( 2 ) ) ( ( 2 2 2 2 2 r R l l r R r e E mr dr r dR r dr d Equation Radiale Les solutions de cette équation sont de la forme : ) ( ) Ζ ( exp ) ( , , 0 r L a n r N r R l n l n Polynôme de Laguerre - L2 Nombre quantique principal Nombre quantique secondaire Donc les fonctions hydrogénoîdes sont des fonctions qui s’écrivent : n,l,m(r,,)=Rn,l(r ) Yl,m(,,)
- 45. 45 Dans l’expression des harmoniques sphériques l,m dépendent de l et m R(r) dépend de n et de l Rn,l D’où n,l,m = Rn,l(r) x l,m() Les valeurs propre associées aux différentes fonctions propres sont : o n a n e Z me x n Z E 2 2 2 2 4 2 2 2 2 ao = 2 2 me ao : c’est le rayon de la première orbite de Bohr En unité atomiques : ћ = 1 ; m = 1 ; e = 1 ao = 1 u.a ao = 0,529167 Å L’énergie dans l’unité atomique est : Atome d’Hydrogène : 2 2 1 n En (u.a) (Hartrée) Système Hydrogénoïde : 2 2 2n Z E n (u.a) 1 u.a = 27,2 ev = 627,2 kcal Fonction du moment cinétique
- 46. 46 Exemples de Fonctions Rn,l (r) : On a toujours -l m l n ≥1 (on l’admet) 0 l n-1 n l m R n,l (r) n,l,m(r,,) En (ev) 1 0 0 2( 2 / 3 ) o a Z e-Zr/ao ao Zr o e a Z / 2 / 3 ) ( 1 -13,6 Z2 2 0 0 2( ) 2 o a Z 3/2 (1- 0 2a Zr )e-Zr/2ao o a Zr o o e a Zr a Z 2 / 2 / 3 ) 2 1 ( ) ( 1 -3,4 Z2 2 1 0 o o a Zr a Z 2 ) ( 3 2 e-Zr/2ao o o a Zr a Z 2 ) ( 3 2 e-Zr/2ao 4 3 cos -3,4 Z2 2 1 1 ’’ o o a Zr a Z 2 ) ( 3 2 e-Zr/2ao 4 3 sin cos ’’ 2 1 -1 ’’ o o a Zr a Z 2 ) ( 3 2 e-Zr/2ao 4 3 sin sin ’’
- 47. 47 II./. Analyse des résultats II.1./- Etat fondamental de l’atome d’hydrogène Atome d’hydrogène : E1 = - 2 1 (u a) = -13,6 (ev) Pour n=1 1 seul fonction 12 Pour n=2 4 valeur de m 4 fonctions 22 dégénérescence d’ordre 22 Donc la dégénérescence est d’ordre n2 Le potentiel d’ionisation de l’atome d’hydrogène est : PI = 13,6 (ev) C’est l’énergie nécessaire pour arracher l’électron de l’atome et l’envoyer à l’infini. Les fonctions hydrogénoïdes sont orthogonales : <n,l,m | n’,l’,m’ > = µnn’ , µl,l’ , µm,m’ Indice de Kronicker
- 48. 48 II.2./- Etude des fonctions d’onde de l’atome d’hydrogène 100 = 1s o a r o e a / 3 1 est de symétrie sphérique Toutes les fonction n,0,0 appelées n,s sont indépendantes de et et donc elles sont de symétrie sphérique. s ao Zr o e a Z / 2 / 3 ) ( 1 Z=1 : atome d’hydrogène La fonction d’onde de l’état fondamental (n=1) est définie comme suit : Pour un système hydrogénoîde 2 : est la densité de probabilité de présence de l’électron dans l’élément de volume d localisé en un point M : Densité ponctuelle: dp(M) d P d M dp 3 2 ) (
- 49. 49 P = |2 r2 sin dr d d = d3P = |2 r2 dr sin d d d3P = ||2d d = r2 sin dr d d ; ||2 = dp (M) P = |2 4 r2 dr = D(r )dr ; D( r): est la densité radiale pour les fonctions n,s D(r )= |ns2 4 r2 4 II.2.1/ Etude de la densité de probabilité ponctuelle pour la fonction n,s : |2 ao r o e a / 2 3 1 Densité de probabilité ponctuelle est aussi de symétrie sphérique o S a r o e a / 3 1 ψ ψ 1 100 est de symétrie sphérique |1S|2 d P d 3 Densité de probabilité ponctuelle
- 50. 50 50 Densité de probabilité ponctuelle La densité ponctuelle est maximale au voisinage immédiat du noyau ,c’est là où on a plus de chance de rencontrer l’électron
- 51. 51 D(r) : est la densité de probabilité de présence de l’électron entre les deux sphères de rayon r et r+dr : Densité Radiale P = ||2 r2 sin dr d d = d3 P = |Rn,l(r)|2 |yl,m(,)|2 r2 sin dr d d = r2 |Rn,l(r)|2 dr |yl,m(,)|2 sin d d = cste r2 |Rn,l(r)|2 dr ao r o e a / 2 3 2 1 r π 4 D(r) ao r o e a r / 2 3 2 4 ao r o e a r / 2 3 2 4 D(r) 3 8 (r) D' o a r ao r e / 2 ) 1 ( o a r II.2.2/ Etude de la densité de probabilité Radiale pour une fonction n,l,m: D(r ) Cste D(r ) P = D(r ) dr o S a r o e a / 3 1 ψ ψ 1 100 est de symétrie sphérique Dans le cas de la fonction 1s de l’atome d’hydrogène :
- 52. 52 Représentation de Densité de probabilité Radiale : 1s; 2s, 2p r2 |Rn,l(r)|2 Densité Radiale r On remarque que D(r ) est nulle au voisinage immédiat du noyau pour les trois fonctions.
- 53. 2 2ème ème Partie Partie 17 Chapitre III Les Méthodes d’Approximations Introduction La résolution analytique de l’équation de schrödinger pour un système hydronénoïde est parfaitement réalisable en faisant une séparation des variables connaissant l’hamiltonien du système . On considère un système simple un noyau autour du quel gravite deux électrons. L’hamiltonien de ce système dans le cadre de l’approximation (B-O-P) s’écrit : 3 2 1 2 1 ˆ ˆ ˆ ˆ ˆ ˆ V V V T T H + +2 e- e1 - e2 - r1 r12 r2
- 54. 18 2 1 2 2 2 1 2 2 1 2 ) ( 2 ˆ r e r Ze r Ze m H ) 2 ( ) 2 ( ˆ 2 2 2 2 1 2 1 2 r Ze m r Ze m H 12 2 2 1 12 2 ˆ ˆ r e H H r e + H’ On ne sait pas résoudre H = E de façon rigoureuse à cause du terme d’interaction (e- , e- ). En effet, il n’existe aucun système de coordonnés qui permet de faire la séparation des variables du terme r12 . Alors pour faire une résolution il faut supprimer le terme d’interaction e- , e- . L’équation de Ĥ n’est jamais résolue pour des systèmes autre que l’hydrogénoïde.
- 55. 19 Au lieu d’obtenir une résolution rigoureuse (impossible). Alors on fait appel à des méthodes d’approximation. Il y on a en nombre de 2 méthodes. Méthode des perturbations Méthode des variations A- Méthode des perturbations I- Cas de non dégénérescence 1°) Exposé de la méthode : on ne traite que le cas de la perturbation indépendante du temps et le cas où on n’a pas de dégénérescence. On va approcher le système à un système dont l’étude est connue.
- 56. 20 Système connus 0 Ĥ o i o i o i E le système dont on Connait la résolution est le système hydrogénoïde. Système inconnue Ĥ i i i E Les 2 systèmes ne doivent pas être très différent c'est-à-dire que : ' ˆ ˆ ˆ H H H o ' Ĥ est faible en grandeur devant 0 Ĥ pour que les 2 systèmes soient équivalents ' Ĥ << 0 Ĥ Ĥ 0 Ĥ ' Ĥ ’ : opérateur de perturbation
- 57. On peut considérer le terme 12 2 r e terme de perturbation P H H o ˆ ˆ : paramètre P̂ : opérateur de perturbation 0 ≤ ≤ 1 vient alors d’un développement en série ; avec P H ' On s’arrête au 1er terme. On écrit : 2 2 1 i i i i E E E E + ……… ......... 2 2 1 i i i i 2 1 , i i E E ce sont des termes de correction pour l’énergie 21
- 58. 23 2°) Perturbation d’ordre 1 a/ Energie ii i i i P P E 1 Remarque : La correction d’ordre 1 sur l’énergie ne fait intervenir que la fonction d’onde d’ordre 0. b/ Fonction propre j j j j i ji j ij i E E P 1 ceci pour ij
- 59. 32 B- Méthode des Variations Basée sur un théorème théorème des variations Théorème des variations : quand on dispose d’un système caractérisé par son hamiltonien et on veut déterminer ces valeurs propres et fonctions propres. Alors 0 / / / E H La valeur moyenne de H <H>/ est supérieure ou égal à une valeur E0 seuil. E0 : c’est la plus petite des valeurs propres de H. Seuil
- 60. 34 Application : on a un système ayant 2 fonctions 1, 2. = C1 1 + C2 2 Théorème des variations 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 / / / C C C C C C H C C E 2 1 2 1 2 2 2 2 1 1 2 1 2 1 2 1 2 2 2 2 1 1 2 1 / 2 / / / / 2 / / / / C C C C H C C H C H C E 12 2 1 2 2 2 1 12 2 1 22 2 2 11 2 1 2 2 C C C C H C C H C H C E L’énergie E est donc fonction de C1, C2 E = f(C1, C2) On cherche le minimum de la fonction f là où s’annule f’. V U E ; 0 ' ' ' 2 V U V V U E E V U V U ' ' H est un opérateur hermitique
- 61. 35 0 2 1 C E C E E C C H C H C 12 2 1 12 2 11 1 2 2 2 2 C1(H11-E)+C2(H12-12E) = 0 E C C H C H C 12 1 2 12 1 22 1 2 2 2 2 C1(H12-12) + C2(H22-E) = 0 S : système homogène n’admet de solution que si le déterminant est nul H11-E H12-E12 = 0 H12- H22-E Si = 0 (base orthogonale) (H11-E)(H22-E) - 2 12 H = 0 H11-E H12 = 0 déterminant séculaire H12 H22-E S
- 62. 38 Généralisation = n n n C n n m n m n n m m n m n n n m m n n n m m n n C C H C C C C C H C E / / / / * * = n m n m m n n m nm m n C C H C C , * * les Cn sont réels 0 m C E n n m n n nm n C H C E , ; n n m nm n E H C 0 ) ( , Cm(Hmm-E) + m n n m n m E H Cn 0 ) ( , , 0 , , n m n m E H déterminant séculaire
- 63. Chapitre IV Atomes à plusieurs électrons Pour un système ayant une charge nucléaire Ze et possédant N électrons, l’Hamiltonien s’écrit (en supposant le noyau fixe : Approx de Born et Oppenheimer) : ij i i r e r ze me H 4 4 2 ˆ 2 2 2 Ĥ = Te + VNe +Vee ; avec Te : énergie cinétique des électrons VNe : potentiel d’interaction noyau- électrons Vee : potentiel d’interaction électron -électron i>j i Énergie cinétique de l’électron Attraction N-e- Répulsion e-_e- : terme positif i Ĥ S’écrit : = hi + Ĥ ij r e 2 i>j i monoéléctronique biéléctronique i i r ze m hi 2 2 2 ; avec Hamiltonien d’un système hydrogénoîde I. Hamiltonien du système H est un hamiltonien électronique : Hel
- 64. • L’atome d’hydrogène et les ions hydrogénoÏdes sont les seuls systèmes atomiques pour lesquels il est possible d’obtenir des fonctions d’onde exactes par résolution directe de l’équation de Schrödinger. • Pour l’atome à n électrons, la difficulté provient du terme biélectronique dans l’expression de H. • Ce terme étant fonction des coordonnées de 2 électrons, la séparation des variables devient impossible quelque soit le système de coordonnée choisi. • Il est donc nécessaire d’utiliser des méthodes d’approximations
- 65. A cause du terme de répulsion électronique, on ne sait pas résoudre l’équation aux valeurs propres d’un atome à plusieurs électrons. Alors on fait appel à des résolutions approchées. Parmi les modèles proposés pour la description de ce système on cite : le modèle à particules indépendantes (MPI), le modèle de Slater (MS) et le modèle de Hartrée-Fock (HF). II. Modèle à particules indépendantes (M.P.I) C’est un modèle dans lequel on néglige toutes les interactions entre les électrons (interactions biélectroniques c.à. d les répulsions) 1 2 Exemple : cas de l’atome d’Hélium = hi Ĥ ; ) 2 ( 2 2 i i r ze m Ĥ = h1 + h2 + e2/r12 h1 : est un hamiltonien hydrogénoîde Soit 1 (1) : fonction propre de h1 : h1 1 (1)= E1 1 (1) Soit 2 (2) : fonction propre de h2 : h2 2 (2)= E2 2 (2) + +2e- r12 r1 r2 0 2 j i r e Hamiltonien Monoélectronique
- 66. La fonction propre de l’hamiltonien H est la fonction décrivant le mouvement des deux électrons : (1,2) = 1 (1)x 2 (2) L’équation aux valeurs propre de l’atome He est : H (1,2) = E (1,2) (h1+ h2) 1 (1)x 2 (2)= E 1 (1)x 2 (2) h1 ne va agir que sur la fonction 1 (1) h2 ne va agir que sur la fonction 2 (2) e1 1 (1)x 2 (2) + e2 1 (1)x 2 (2) = E 1 (1)x 2 (2) (e1 + e2) 1 (1)x 2 (2) = E 1 (1)x 2 (2) E = (e1 + e2) L’énergie du système est la somme des énergies monoélectroniques • Généralisation: Pour un système à n électrons H= hi (dans le modèle à électrons indépendants). * La fonction d’onde est : (1,2,3,…..n) = x i(i) fonction monoéléctronique * l’énergie est : E = ei ; avec (H) 1 2 2 e n z ei
- 67. Les fonctions d'onde monoélectroniques i(i) sont appellées orbitales atomiques, et les valeurs propres sont appellées énergies orbitalaires «ei». Si l'on joint à l'orbitale une fonction de spin pour le i-ème électron, alors on obtient une spin-orbitale, par exemple. Il est important de bien distinguer l’orbitale, qui est une fonction d'onde pour un et un seul des électrons du système, de la fonction d'onde de l'atome ou de la molécule en entier, qui est un produit de n orbitales ou spin-orbitales. III. Principe d’indiscernabilité 1/- Statistique de Fermi- Dirac D’après cette statistique : les électrons sont des particules à spin demi entier (Fermions) qui ne peuvent être séparés ou individualisés : ce sont des particules indiscernables. La fonction d’onde d’un système doit être antisymétrique par rapport à l’échange de deux particules. Ex: Cas de 2 e- de He : la fonction d’onde qui décrit le système est : He : (1,2) = - (2,1) 1 2
- 68. 2/- Principe de Pauli Deux électrons ne peuvent jamais avoir leur nombres quantiques identiques. Ils différent aux moins par le spin. : (1,2) = 1s (1) (1). 1s (2) (2) ; les 2 fonctions ne différent 1 2 que par le spin même n, l, m 3/- Antisymétrisation des fonctions d’onde La fonction d’onde antisymétrique qui tient compte de l’échange entre les 2 électrons de He ( toutes les permutations) s’écrit : déterminant de Slater Généralisation U1(1) U2(1) U1(1) U2(1)….. Un(1) ’ (1,2)= ; ’ (1,2,3,…….n) = U1(2) U2(2) U1(2) U2(2)….. Un(2) U1(n) U2(n)….. Un(n) Spin- orbitale U1 U2 2 1 ! 1 n
- 69. Remarque: l’antisymétrisation de la fonction, dans le cas de plusieurs électrons n’apporte aucune amélioration à l’énergie du système si l’hamiltonien est du type mono- électronique. 4/- Analyse des Résultats • Cas de l’atome He : E = e1+e2 = -4 u.a= -108,8 ev Hélium - 108,8 - 78,6 II. Modèle des constantes d’écran : Modèle de Slater 1/- Principe : Il s’agit d’un modèle électrostatique qui consiste à introduire la répulsion entre électrons en diminuant la charge du noyau que subit un électron donné. + x x x x Effet Ecran: Z Z*= Z - écran e- +Ze C/C : M.P.I est un modèle grossier qu’il faut améliorer Grand écart
- 70. L’hamiltonien d’un atome à plusieurs électrons dans l’approximation de Slater est de type mono-électronique donné par : eff eff i i i m h ) (r) V 2 ( H 2 i i 2 i i 2r -1) * (n * n r * Z - (r) V eff i ; Z* = Z - écran Potentiel Effectif Constante d’écran n 1 2 3 4 5 6 n* 1 2 3 3,7 4 4,2 Hamiltonien qui dépend uniquement des cordonnées de l’électron i : mono électronique Les constantes d’écran σ sont calculées de la façon suivante: • Les orbitales sont classées en groupe de Slater: • (1s) (2s2p) (3s3p) (3d) (4s4p) (4d) (4f) (5s5p)….
- 71. Électron j/électron i 1s 2s 2p 3s 3p 3d 4s 4p 4d 1s 0,30 2s 2p 0,85 0,35 3s 3p 1 0,85 0,35 3d 1 1 1 0,35 4s 4p 1 1 0,85 0,85 0,35 4d 1 1 1 1 1 0,35 2 /- Energie et fonctions d’onde de Slater * L’énergie d’un électron « i » dans le cadre du modèle de Slater est : ) ( * 1 2 2 H Ε n Z* Ε i E1 (H) = -13,6 ev = - 0,5 u.a : énergie de l’état fondamental de l’atome d’hydrogène
- 72. ) * n r * Z xp(- 1 * r N R(r) e n • La fonction d’onde est donnée par : n,l,m (r,, ) = R(r) Yl,m (, ) • La forme de la fonction de Slater est en relation étroite avec celle des systèmes hydrogénoîdes. En effet, Slater a conservé la partie angulaire et a modifié uniquement la partie radiale. • en u.a • Les fonctions de Slater ne constituent pas une base orthogonales. Mais celles –ci sont mieux adaptées à décrire l’atome que les orbitales hydrogénoïdes. 3 /- Analyse des résultats Atome Modèle Slater (ev) Expérience (ev) He -78 -78,6 Li -204 -203 C -1024 -1025 N -1480 -1486 Modèle Assez Bon pour étudier un Atome à électrons
- 73. La méthode de Hartrée-Fock ou encore méthode de champ auto- cohérant proposé par D. Hartrée consiste à ce que l’interaction de chaque électron de l’atome avec tous les autres est remplacée par l’interaction avec un champ moyen crée par le noyau et la totalités des autres électrons. Cela permet de remplacer le potentiel de type (1/rij) que l’on trouve dans l’Hamiltonien et qui dépend des coordonnées des deux électrons, par une expression définissant l’interaction électronique en tant qu’une fonction des coordonnées de chaque électron isolé. III. Modèle de Hartrée-Fock
- 74. III.1. Position du problème L’approximation de Born-Oppenheimer nous amène à chercher des solutions (approchées) de l’hamiltonien électronique (en unités atomiques) : ) , ( ˆ ) ( ˆ 4 4 2 ˆ 2 2 2 j i H i H r e r ze me H ij i i el Les deux premiers termes font intervenir les coordonnées spatiales d’un seul électron i, il s’agit d’un opérateur monoélectronique noté : Le troisième terme fait intervenir les coordonnées de deux électrons i et j, C’est un opérateur biélectronique. Il peut être noté : ) ( ˆ i H ) , ( ˆ j i H Puisque : rij = |ri – rj|
- 75. III.2 Détermination de l’énergie L’énergie moyenne associée à un déterminant de Slater (DS) normalisé est : Le calcul dans le cas général apparaît assez complexe, puisque le bra et le ket contiennent le n ! permutations des coordonnées de n électrons. Le résultat est heureusement simplifié par l’orthonormalité des spin- orbitales qui entraîne la nullité d’un grand nombre de termes. < U1(1)-----Un(n) U1(1)-----Un(n) > ! 1 n ! 1 n el Ĥ III.2.1. Système à deux électrons L’hamiltonien se réduit, avec les notations précédentes à: 12 1 ) 2 ( ˆ ) 1 ( ˆ ˆ r H H el H et la fonction d’onde s’exprime en fonction de deux spin-orbitales orthonormales U1 et U2 :
- 76. (1,2)= ! 2 1 U1(1) U2(1) U1(2) U2(2) = 2 2 [U1(1) U2(2) - U2(1) U1(2)] L’énergie se décompose en intégrales monoélectroniques et biélectroniques : E= < | | > = < | | > + < | | > + < | | > 12 1 r el Ĥ ) 1 ( Ĥ ) 2 ( Ĥ = E1 + E2 + J12 - K12 avec E1 et E2 les énergies monoélectroniques J12 et K12 sont des intégrales biélectroniques telles que : J12 = < U1(1) U2(2) | | U1(1) U2(2) > = < 12|12> : Intégrale Coulombienne K12 = < U1(1) U2(2)| | U2(1) U1(2) > = < 12|21> : Intégrale d’échange 12 1 r 12 1 r
- 77. III. 2.2. Cas général Dans le cas d’un système atomique à 2n électrons telles que les couches sont complètes, l’énergie moyenne totale est données par : E = 2 E i - ( 2 J ij –K ij) i=1 n i=1 j=1 n n J ij : intégrale coulombienne K ij : intégrale d’échange
- 78. I. Introduction Un terme spectral ou terme atomique est un état énergétique déterminé de l’atome. Une configuration électronique correspond à un état énergétique de l’atome. Chaque état d’énergie atomique peut être représenté symboliquement par un terme spectral. II. Description des termes énergétiques II.1/ Notation Les niveaux d’énergie sont désignés symboliquement de la façon suivante : Chapitre V LES TERMES SPECTRAUX 38
- 79. où X indique la valeur de L avec la correspondance suivante : L : 0 1 2 3 4 5 X : S P D F G H II.2°/ Moment orbital, moment de spin d’un atome Soit L et S les moments cinétiques d’un atome: L : est le moment cinétique orbital total de l’atome S: est le moment cinétique total de spin de l’atome L et S se couplent à leur tour pour générer un vecteur supplémentaire J (couplage Russell-Saunders): J = L + S ; avec MJ = ML + MS |L-S| J L+S avec J: est le nombre quantique du moment cinétique total 39
- 80. III.Atome sans interaction Si on ne tient pas compte du couplage spin-orbite (L, S), un état énergétique est symbolisé par : 2S+1X. Par effet de l'interaction spin- orbite, ce terme se sépare en autant de niveaux qu’il y a de valeurs de J ; chacun caractérisé par une valeur bien définie de J, comprise entre (L+S) et |L - S|. Cette séparation est appelée structure fine. Le couplage spin-orbite lève donc la dégénérescence du terme 2S+1X. (2s+1) représente la multiplicité de l’état considéré. Celle-ci désigne l’ordre de dégénérescence de l’état. 40
- 81. µ=2s+1 Exemple : S =1 ; 2s+1 = 3 état : 3P ; la dégénérescence de L =1 l’état est : (2S + 1) (2L + 1) = (2x1+1) (2x1+1) = 9 811
- 82. Cet état correspond par exemple à : 2 e- Non apparié tel que : s = 1/2 + 1/2 et L=1+0 IV. Atome avec interaction : Structure Fine Les deux moments cinétiques de l’atome, orbital et de spin, ne sont pas indépendants l’un de l’autre, chacun étant associé à son propre moment magnétique. L’interaction des champs magnétiques engendrés par ces moments est dite: interaction spin-orbite ou (couplage spin-orbite) Cette interaction est à l’origine de la structure fine des spectres atomiques. En particulier la structure à doublet des spectres des métaux alcalins. l =1 l =0 42
- 83. Exemple : 1/ l=1 => J= 0,1,2 => 3P0, 3P1, 3P2 s=1 l - s J l + s 3P 3P0 3P1 3P2 On a éclatement de l’état 3P en 3 états , il s’agit d’un levé de dégénérescence partielle: c’est la structure Fine. 2/ l=2 => J= ½, 3/2, 5/2, 7/2 => 4D½ , 4D3/2 ,4D5/2 ,D7/2 s=3/2 l - s J l + s 3/ * Atome d’hydrogène : 1 seul électron : l=0 => s= ½ 2S½ 2 possibilités pour la position du spin => 2 fonctions d’onde. 43
- 84. 4/ * Atome d’hélium : 2 électrons ml1 = 0; ml2= 0 => ML = 0 => L= 0 ms1 = ½ et ms2=- ½ Ms = 0 => S=0 = terme 1S0 Remarque: Les sous couches pleines n’interviennent pas ,elles sont définies par ML et MS nulles. 5/ * Atome de Bore : 1s2 2s2 2p1 l=1 & s= ½ 2P3/2 ; 2P1/2 => 2 états 6/ * Spectre des métaux alcalins: Ex: Na(Z=11) : 1s2 2s2 2p6 3s1 l=0 et s= ½ 2S1/2 Si on excite l’électron de valence vers un état supérieur : 1s2 2s2 2p6 3p1 l= 1 et s= ½ les termes : 2P3/2 ; 2P1/2. 44
- 85. Remarques : a- l’interaction spin orbite fournit une petite perturbation permettant de levé la dégénérescence partiellement: c’est la structure fine . b- Chaque niveau fin est dégénéré (2j+1) fois. Cette dégénérescence est levé grâce à une forte perturbation tel que le champ magnétique. 2S 2P 2S1/2 2P3/2 2P1/2 1s2 2s2 2p6 3s1 1s2 2s2 2p6 3p1 On constate que chaque raie spectrale se décompose en deux raies très voisines . Cet effet est connu en spectroscopie atomique par : la structure à doublet des spectres des métaux alcalins. 5890 Å 5896 Å 45
- 86. 7/ Terme spectraux de l’atome de carbone : Tableau des micro-états 6C : 1s2 2s2 2p2 : ; il s’agit dans ce cas de 2 électrons état fondamental équivalents (même sous- couche). -Le terme spectral de l’état fondamental est : 3P déterminer directement. -Pour déterminer tous les états correspondant à une configuration, on construit un tableau des micro-états. Ms = 0,1,-1 ; pour Ml= mli = 2,1,0,-1,-2 1 0 -1 2 1 1 1 1 1 1 1 1 0 1 0 1 0 1 0 0 1 -1 1 -1, 1 -1 0 0 1 -1 -1 -1 0 -1 0 -1 0 -1 0 -2 -1 -1 -1 -1 -1 -1 Ms ML + + 1 0 -1 + - 46
- 87. • 1 1 : Ml = 2 ; Ms = 0 => L=2 et S= 0 1D2 dégénérescence =( 2x2+1) • = 5 états • Ml = 1 ; Ms = 1 => L=1 et S= 1 => 3P0,1,2 dégénérescence = ( 2x1+1)x • (2x1+1) = 9 états • Ml = 0 ; Ms = 0 => L=0 et S= 0 => 1s0 1 seul état : 0 0 + - 8/ Terme spectraux de l’atome de carbone dans son état excité : 2p13p1 : cas d’électrons non équivalent ML = ml1 + ml2= 0 + 1 =1 = 0 + (-1) = -1 = 0 + 0 = 0 L= 1 MS = ms1 + ms2= ½ + ½ = 1 = ½ + (- ½) = 0 = (-½) + ½ = 0 = (-½) + (- ½) = -1 S = 1 & S = 0 Les termes spectraux dans ce cas sont déterminés en combinant chaque valeur de L avec toute les valeurs de S 47
- 88. Règles pour classer les états d’une configuration électronique donné: 3 règles: Règles de Hund 1/- L’état fondamental est toujours celui qui correspond à la multiplicité maximale. 2/- Lorsque deux états ont la même multiplicité de spin l’état le plus stable est celui qui correspond à la plus grande valeur de L. 3/- Lorsque la sous- couche est moins que moitié remplit. Le niveau le plus stable est celui qui correspond à la plus petite valeur de J. Et c’est l’inverse lorsque la sous-couche est plus que moitié remplit. 48
- 89. V. Effet ZEEMAN : Structure hyperfine 1/- Définition : On appelle effet Zeeman la modification d’un spectre par l’application d’un champ magnétique. Cette modification se traduit par la décomposition de certaine raie spectrale. 1 seule raie Sans Champ 3 raies Avec Champ 2/- Moment magnétique d’un atome : Il résulte de l’interaction avec le champ magnétique B des moments magnétiques résultants des grandeurs vectoriels caractéristiques de l’atome à savoir les moments cinétiques orbitales et de spins. 49
- 90. Les moments magnétiques de l’atome sont définis par : L - L 2 - B e L m e ; est le magnéton de Bohr e 2 µ m e S 2 - S - B e S m e atome = - B ( L + 2 S ) : est le moment totale de l’atome 3/- Variation de l’énergie d’un atome en présence de B L’ interaction entre le moment totale de l’atome atome et le champ magnétique B , conduit à une variation de l’énergie de l’atome de l’ordre de : E = B g . MJ . B ; avec : MJ : projection de J sur la direction Oz g : est le facteur de Landé ou de décomposition spectrale 1) (J J 2 ) 1 ( ) 1 ( ) 1 ( 1 g S S L L J J 50
- 91. 4/- Détermination de la fréquence E = h E1 = E01 + B gJ1 . MJ1 .B E2 = E02 + B gJ2 . MJ2 . B le E correspondant à l’absorption de la lumière est : E = E2 – E1 = h E = E02 – E01 + B .B (gJ2 . MJ2 – gJ1 . MJ1 ) = h D’où la fréquence : = 0 + (gJ2 . MJ2 – gJ1 . MJ1 ) C’est l’effet Zeeman Anormal h0 h B 51
- 92. Exemple : 1/- Atome d’hydrogène: conf : 1s1 : L=0 , S= ½ ,J = ½ g = 2 E = B .2. (-1/2 ) . B = - B . B E0 E0 + B . B E0 - B . B 2B . B Si on néglige le spin (S= 0) alors gJ2 = gJ1 = 1 : C’est l’effet Zeeman normal = 0 + MJ Les transitions possibles sont tracées en utilisant les règles de sélection h B 52