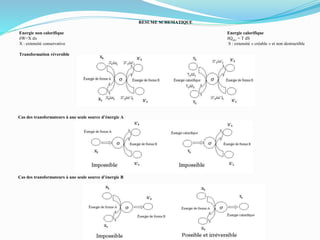
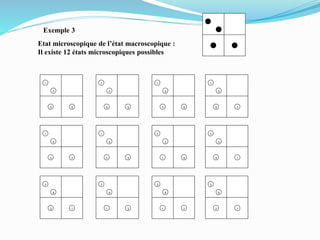
Le document présente une introduction exhaustive à la thermodynamique, décrivant l'évolution historique des concepts de chaleur, de température, et des gaz. Il explique les théories des gaz parfaits et réels, ainsi que les principes des machines thermiques et des transformations énergétiques. Enfin, il aborde les états d'équilibre et les variables d'état nécessaires à l'analyse thermodynamique des systèmes.

























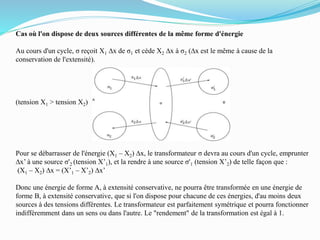















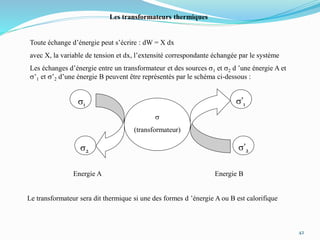

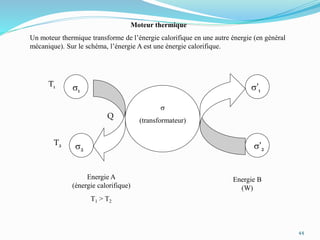
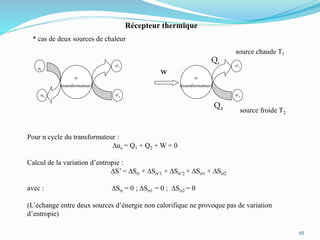




























































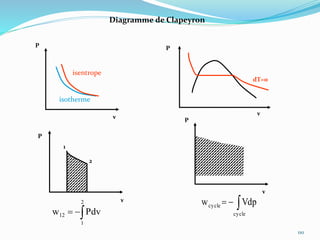
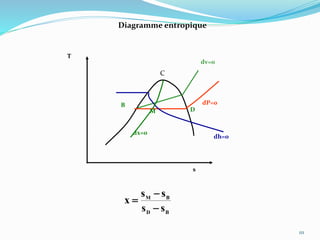



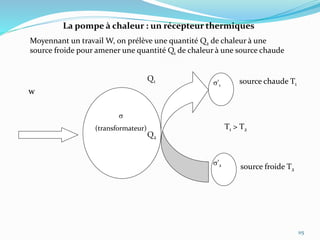




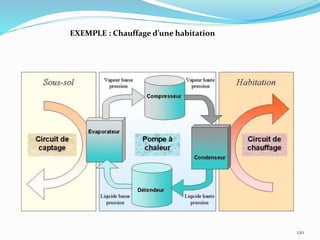


































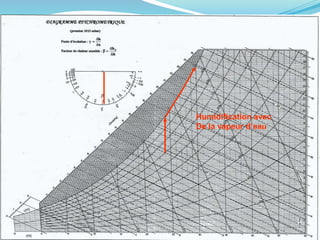


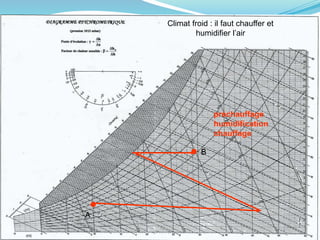

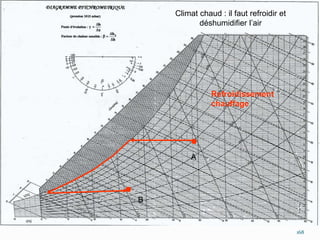























































![[2]Chapitre I.1 Biophysique de la respiration 2021.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/2chapitrei-240825231633-dcea161f-thumbnail.jpg?width=640&height=640&fit=bounds)











