Téléchargé 134 fois









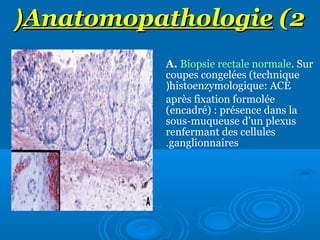































Le document traite de la maladie de Hirschsprung, une affection rare caractérisée par l'absence de cellules ganglionnaires dans l'intestin, menant à des occlusions intestinales chez les enfants. Il décrit son épidémiologie, les méthodes de diagnostic, les techniques chirurgicales modernes pour le traitement ainsi que la voie génétique potentielle de cette maladie. En conclusion, un diagnostic précoce est essentiel pour prévenir des complications graves, et les avancées chirurgicales ont amélioré la prise en charge des patients.










![Kyste hydatique du foie [enregistrement automatique]](https://cdn.slidesharecdn.com/ss_thumbnails/kystehydatiquedufoieenregistrementautomatique-170224222408-thumbnail.jpg?width=640&height=640&fit=bounds)



















































