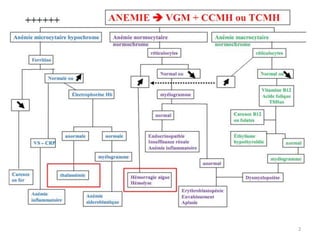
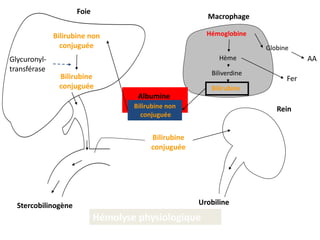
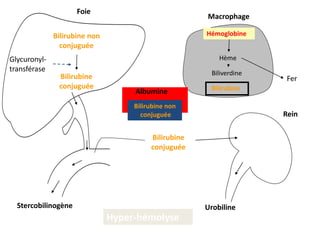
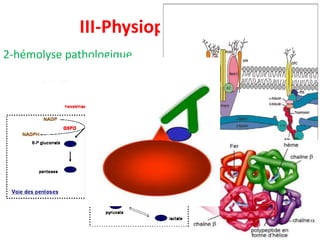
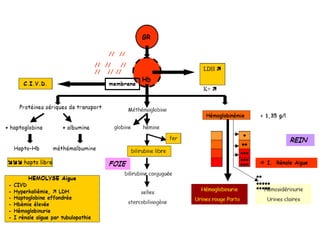
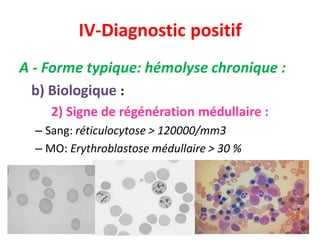
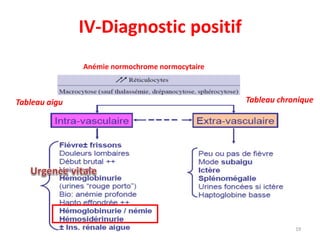
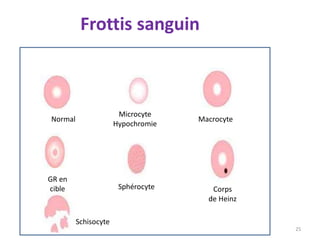
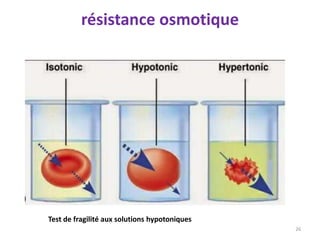
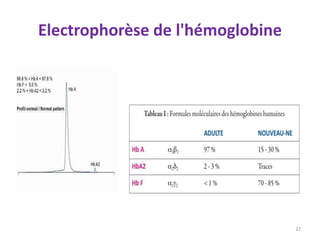
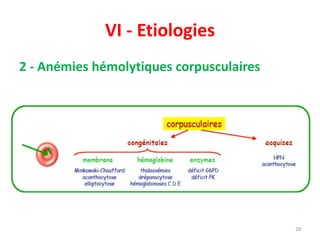
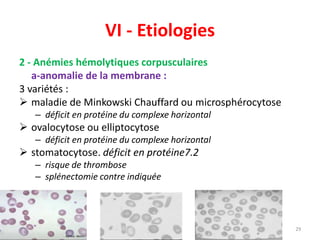
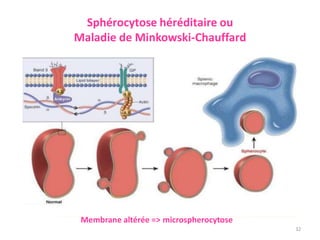
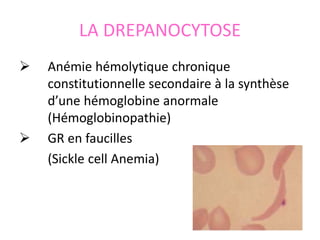
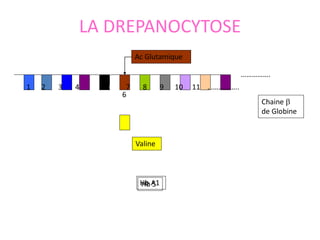
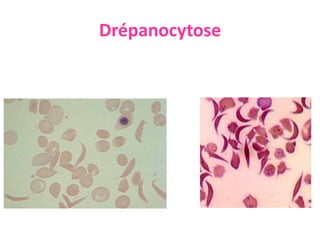
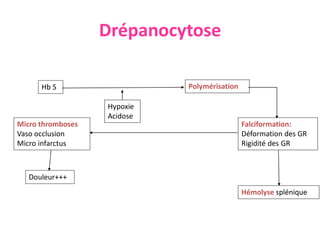
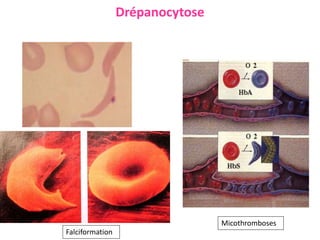
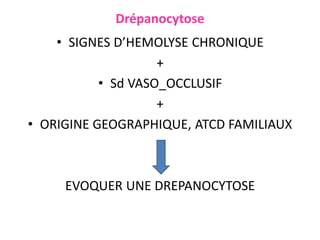
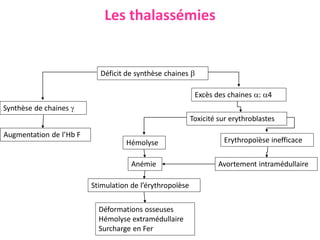
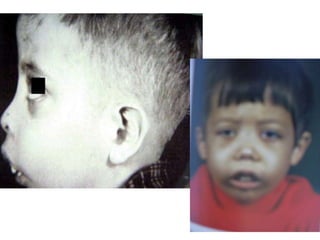
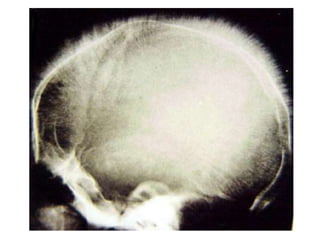
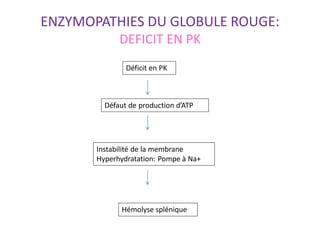
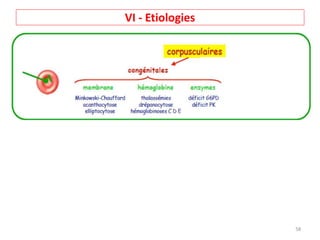
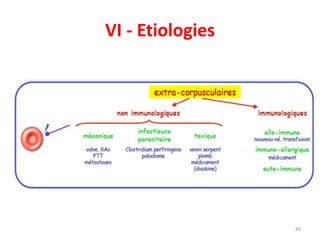
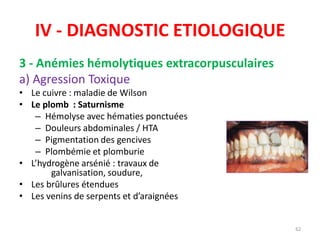
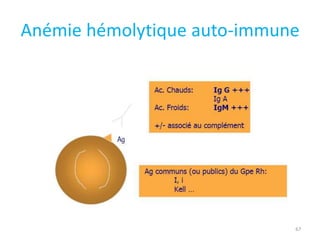
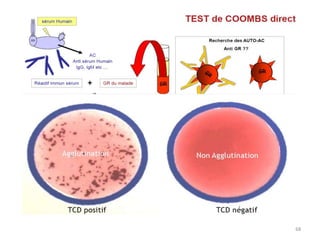
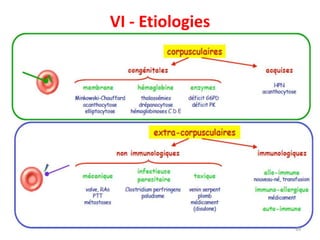
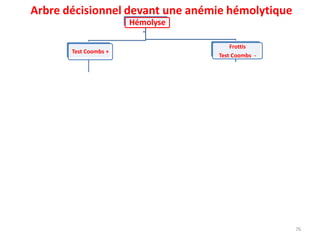
Le document traite des anémies hémolytiques, en définissant les concepts clés et en présentant un plan de cours détaillé concernant la classification, la physiopathologie, le diagnostic et le traitement. Il aborde également les étiologies des anémies hémolytiques, y compris les anomalies des globules rouges et les facteurs externes impliqués. Enfin, le document souligne l'importance du diagnostic différentiel pour poser un diagnostic positif.