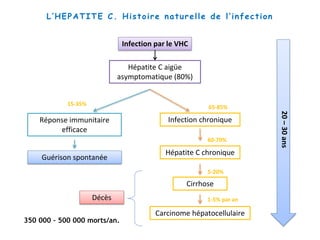
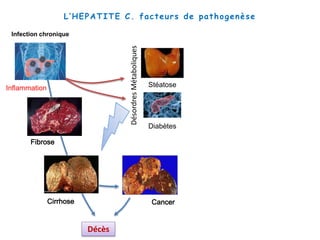
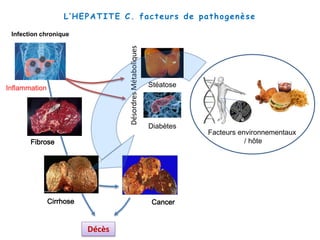
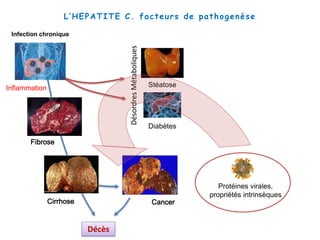
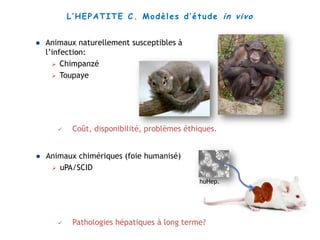
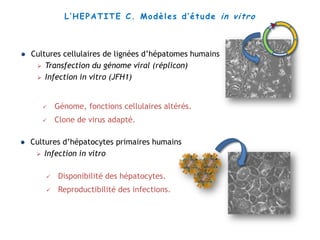
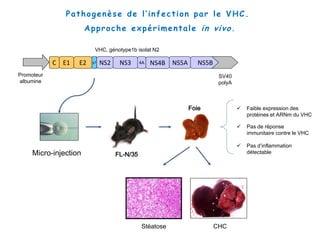
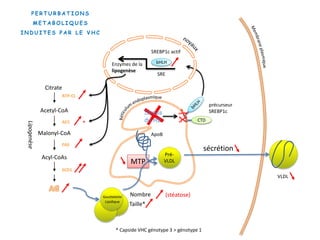
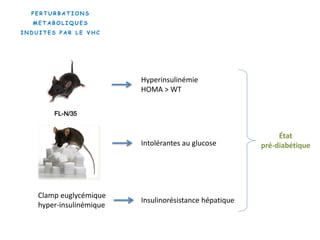
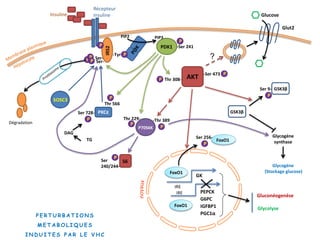
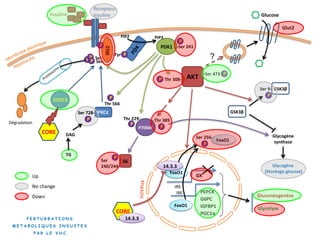
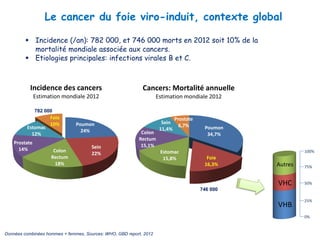
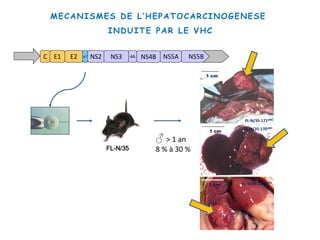
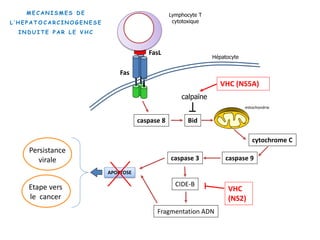
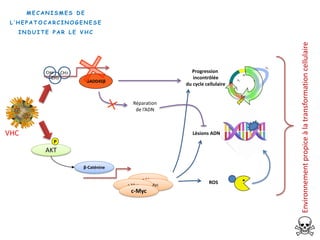
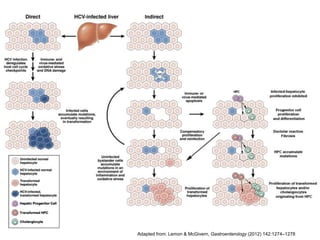
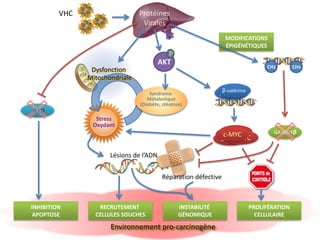
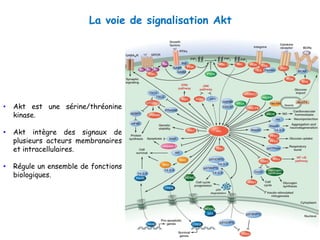
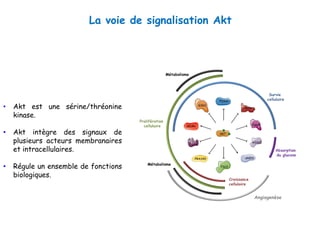
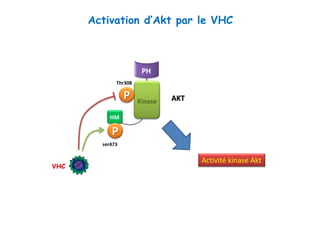
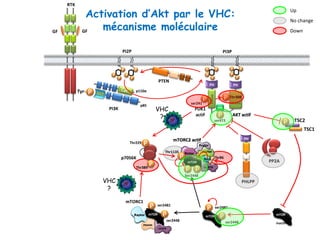
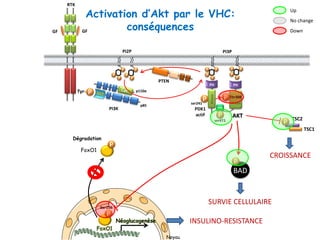
Le document explore la pathogenèse des lésions hépatiques causées par le virus de l'hépatite C (VHC), en détaillant les mécanismes moléculaires et les facteurs de risque associés à l'infection chronique, y compris le développement de la cirrhose et du cancer hépatocellulaire. Il souligne l'influence des protéines virales sur les fonctions cellulaires et décrit les approches expérimentales utilisées pour étudier ces mécanismes, ainsi que les perturbations métaboliques qu'elles induisent. En outre, il aborde la fréquence élevée des cas de cancer du foie liés à cette infection et la nécessité de recherches supplémentaires pour comprendre pleinement les interactions entre le VHC et les processus cancéreux.