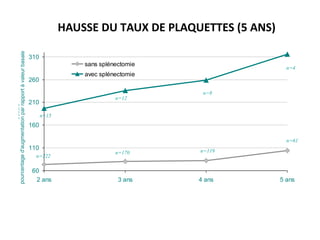
Le document traite des maladies lysosomales, mettant en évidence leur classification, caractéristiques et traitements disponibles. Des détails sur des maladies spécifiques comme la maladie de Gaucher et la maladie de Fabry sont fournis, ainsi que des informations sur l'évolution des traitements, notamment l'enzymothérapie substitutive. Une attention particulière est portée sur les défis et limites des thérapies actuelles, ainsi que sur l'importance d'un suivi clinique régulier.