Téléchargé 45 fois







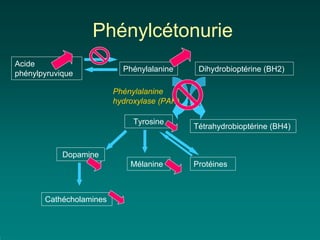












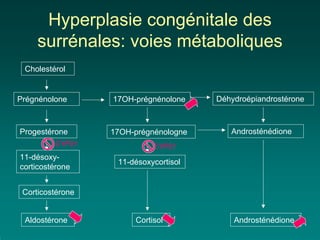



Le dépistage néonatal en France cible cinq maladies depuis 2003, dont la phénylcétonurie et l'hypothyroïdie congénitale, avec des méthodes de prélèvement sanguin précoces et un taux de couverture supérieur à 99%. La phénylcétonurie, une maladie métabolique, requiert un régime strict dès la naissance pour éviter des séquelles sévères, tandis que l'hypothyroïdie congénitale nécessite un traitement précoce par l-thyroxine pour un pronostic favorable. Les dépistages visent à identifier rapidement ces conditions pour permettre des interventions précoces et prévenir des handicaps irréversibles.



























































![Comas 3eme année [lecture seule]](https://cdn.slidesharecdn.com/ss_thumbnails/comas3emeannelectureseule-130912063627-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)







