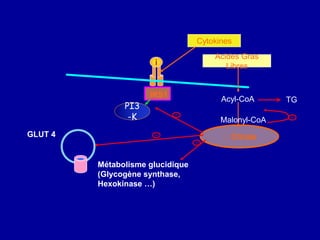
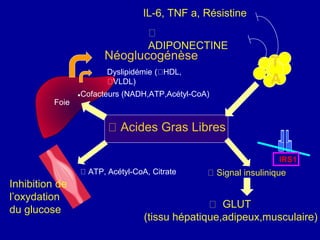
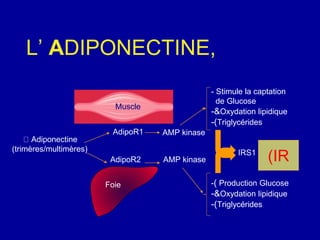
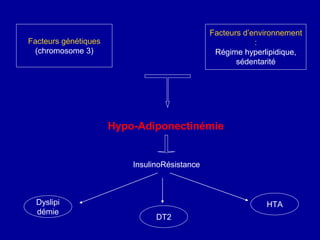
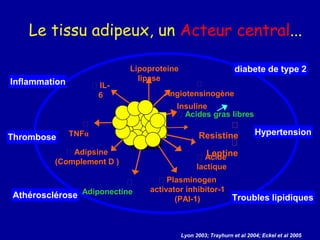
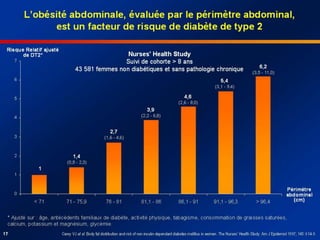
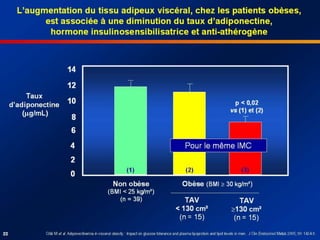
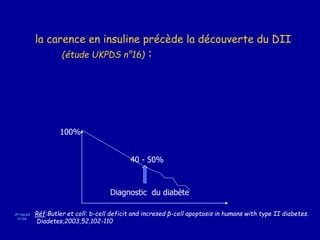
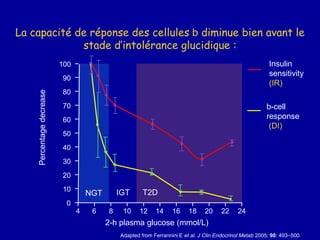
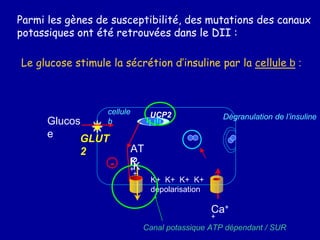
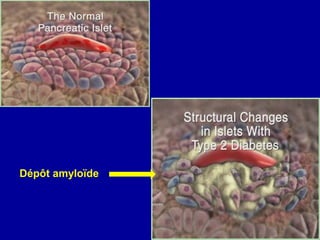
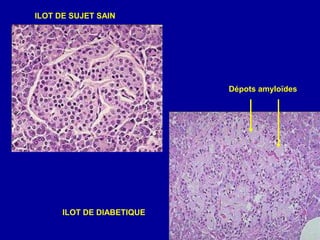
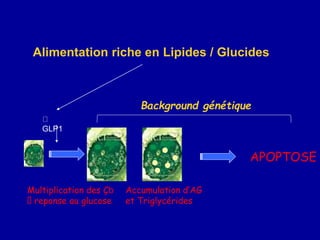
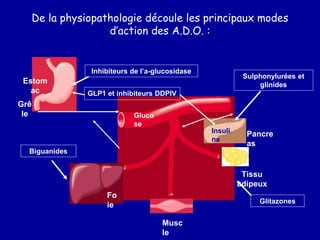
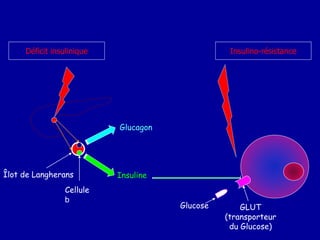
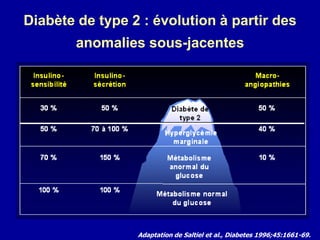
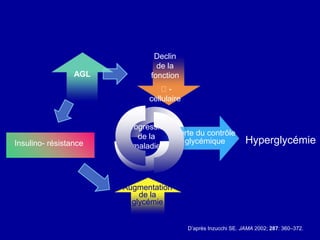
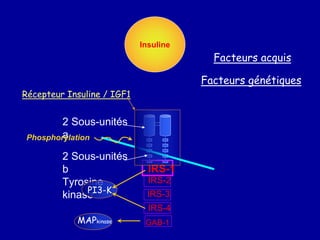
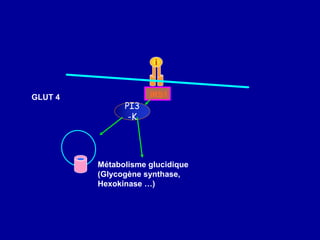
Le diabète de type 2 est caractérisé par une insulinorésistance et un dysfonctionnement des cellules bêta, entraînant une hyperglycémie progressive et une perte de contrôle glycémique. Les facteurs contribuant à cette pathologie incluent des éléments génétiques, environnementaux et physiologiques, ainsi que des anomalies dans la sécrétion d'insuline et de glucagon. La gestion de cette maladie nécessite une compréhension approfondie de la physiopathologie sous-jacente, impliquant des mécanismes complexes d'interaction hormonale et métabolique.











![● Randle (1963) :
In vitro : m captation du Glucose en présence d’AGL en
excès. compétition ?
en fait, défaut de GLUT4 en surface
● k [AGL] sur tout le nycthémère dans le DII (Reaven).
● Corrélation forte entre IR et contenu intramyocytaire en
triglycérides : X 6 dans DII
Réf: Acides gras et résistance à l’insuline – J. GIRARD
Métabolismes Hormones Diabètes et Nutrition (VIII),n°1,janv.fév.2004, p14-20](https://image.slidesharecdn.com/physiopathdt2hhtouggortfinal2-150626020715-lva1-app6892/85/Dr-Hitache-DT2-PHYSIOPATHOLOGIE-16-320.jpg)